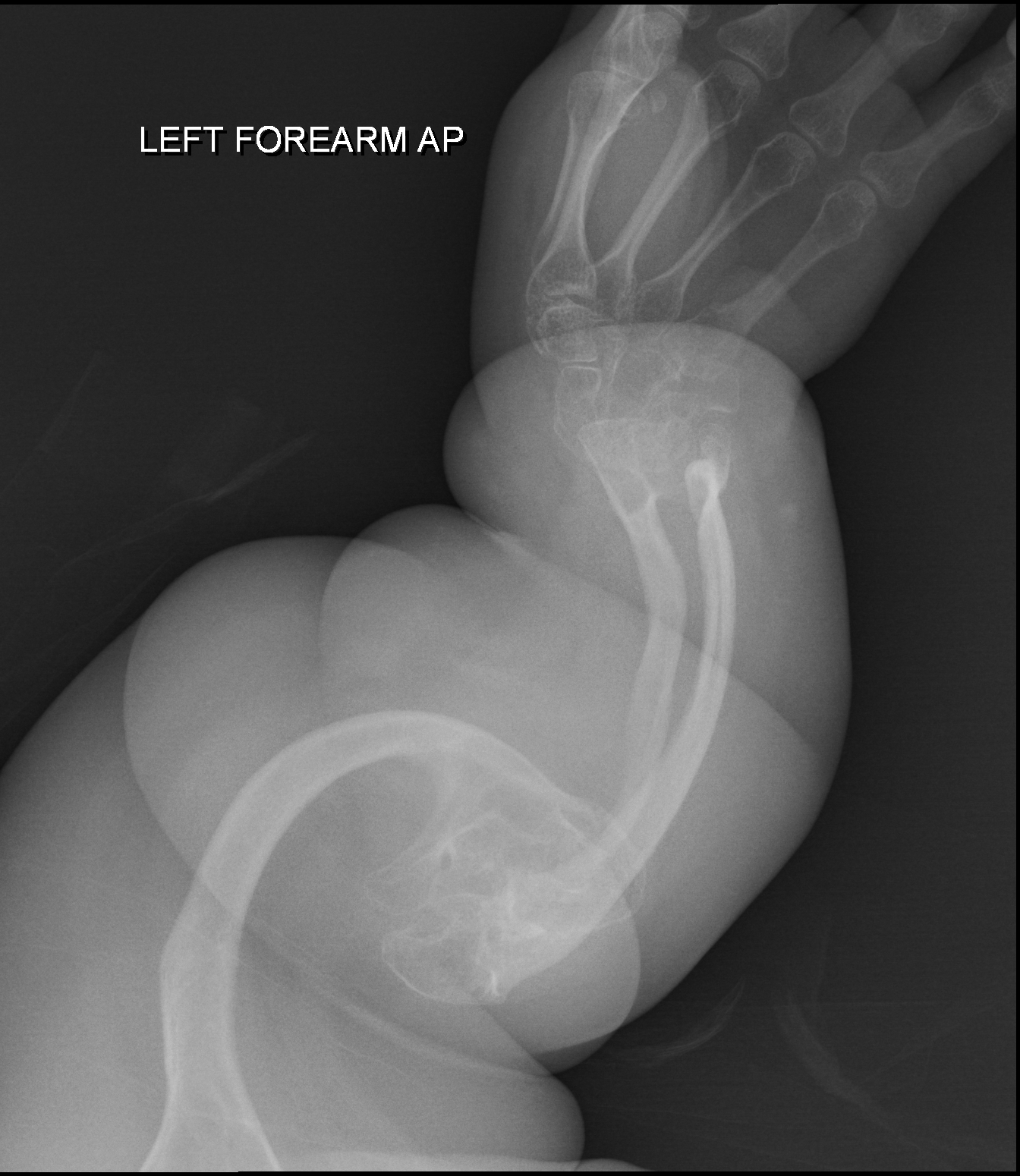
Uma displasia óssea grave, que leva à morte no período perinatal (próximo ao nascimento), caracterizada por encurtamento dos membros (braços e pernas), crânio de tamanho normal com fenda no céu da boca, polegares que se dobram para trás de forma incomum ("polegares de carona"), características faciais marcantes e alterações nos ossos visíveis em exames de raio-X. É causada por mutações no gene do transportador de sulfato da displasia distrófica.
Introdução
O que você precisa saber de cara
Uma displasia óssea grave, que leva à morte no período perinatal (próximo ao nascimento), caracterizada por encurtamento dos membros (braços e pernas), crânio de tamanho normal com fenda no céu da boca, polegares que se dobram para trás de forma incomum ("polegares de carona"), características faciais marcantes e alterações nos ossos visíveis em exames de raio-X. É causada por mutações no gene do transportador de sulfato da displasia distrófica.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 30 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 72 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisSulfate transporter which mediates sulfate uptake into chondrocytes in order to maintain adequate sulfation of proteoglycans which is needed for cartilage development (PubMed:11448940, PubMed:15294877, PubMed:20219950, PubMed:7923357). Mediates electroneutral anion exchange of sulfate ions for oxalate ions and of sulfate and oxalate ions for chloride ions (PubMed:20219950). Mediates exchange of sulfate and oxalate ions for hydroxyl ions and of chloride ions for bromide, iodide and nitrate ions (
Cell membraneApical cell membrane
Diastrophic dysplasia
An autosomal recessive disease characterized by osteochondrodysplasia with clinical features including dwarfism, spinal deformation, and specific joint abnormalities.
Variantes genéticas (ClinVar)
272 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 736 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
3 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Atelosteogênese II
Centros de Referência SUS
24 centros habilitados pelo SUS para Atelosteogênese II
Centros para Atelosteogênese II
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Computational biology insights into genotype-clinical phenotype-protein phenotype relationships between novel SLC26A2 variants identified in inherited skeletal dysplasias.
Pathogenic variants in the transmembrane sulfate transporter protein SLC26A2 are associated with different phenotypes of inherited chondrodysplasias. As limited data is published from India, in this study we sought to elucidate the molecular basis of inherited chondrodysplasias in an Indian cohort. Molecular screening of 32 fetuses with antenatally diagnosed lethal skeletal dysplasia was performed by next generation sequencing and Sanger sequencing. The genotype-protein phenotype characterization was done using computational biology techniques like homology modelling, stability and pathogenicity predictions. We identified five rare autosomal recessive SLC26A2 [NM_000112.4] variants, including three homozygous c.796dupA(p.Thr266Asnfs*12), c.1724delA(p.Lys575Serfs*10), and c.1375_1377dup(p.Val459dup) and two heterozygous variants (c.532C > T(p.Arg178*)) and (c.1382C > T(p.Ala461Val)) in compound heterozygous form in a total of four foetuses. Genotype-protein phenotype annotations highlighted that the clinically severe achondrogenesis 1B causative c.796dupA(p.Thr266Asnfs*12) and c.1724delA(p.Lys575Serfs*10)variants impact SLC26A2 protein structure by deletion of the protein core and transmembrane STAS domains, respectively. In clinically moderate atelosteogenesis type 2 phenotype, the c.1382C > T(p.Ala461Val) variant is predicted to distort alpha helix conformation and alter the bonding properties and free energy dynamics of transmembrane domains and the c.532C > T(p.Arg178*) variant results in loss of both core transmembrane and STAS domains of the SLC26A2 protein. The c.1375_1377dup(p.Val459dup) variant identified in clinically milder atelosteogenesis type II-diastrophic dysplasia spectrum lethal phenotype is predicted to decrease the Qualitative Model Energy Analysis (QMean), which affects major geometrical aspects of the SLC26A2 protein structure. We expand the spectrum of SLC26A2 related lethal chondrodysplasia and report three novel variants correlating clinical severity and protein phenotype within the lethal spectrum of this rare dysplasia. We demonstrate the relevance of structural characterization to aid novel variant reclassification to provide better prenatal management and reproductive options to families with lethal antenatal skeletal disorder.
Suppressing UPR-dependent overactivation of FGFR3 signaling ameliorates SLC26A2-deficient chondrodysplasias.
Mutations in the SLC26A2 gene cause a spectrum of currently incurable human chondrodysplasias. However, genotype-phenotype relationships of SLC26A2-deficient chondrodysplasias are still perplexing and thus stunt therapeutic development. To investigate the causative role of SLC26A2 deficiency in chondrodysplasias and confirm its skeleton-specific pathology, we generated and analyzed slc26a2-/- and Col2a1-Cre; slc26a2fl/fl mice. The therapeutic effect of NVP-BGJ398, an FGFR inhibitor, was tested with both explant cultures and timed pregnant females. Two lethal forms of human SLC26A2-related chondrodysplasias, achondrogenesis type IB (ACG1B) and atelosteogenesis type II (AO2), are phenocopied by slc26a2-/- mice. Unexpectedly, slc26a2-/- chondrocytes are defective for collagen secretion, exhibiting intracellular retention and compromised extracellular deposition of ColII and ColIX. As a consequence, the ATF6 arm of the unfolded protein response (UPR) is preferentially triggered to overactivate FGFR3 signaling by inducing excessive FGFR3 in slc26a2-/- chondrocytes. Consistently, suppressing FGFR3 signaling by blocking either FGFR3 or phosphorylation of the downstream effector favors the recovery of slc26a2-/- cartilage cultures from impaired growth and unbalanced cell proliferation and apoptosis. Moreover, administration of an FGFR inhibitor to pregnant females shows therapeutic effects on pathological features in slc26a2-/- newborns. Finally, we confirm the skeleton-specific lethality and pathology of global SLC26A2 deletion through analyzing the Col2a1-Cre; slc26a2fl/fl mouse line. Our study unveils a previously unrecognized pathogenic mechanism underlying ACG1B and AO2, and supports suppression of FGFR3 signaling as a promising therapeutic approach for SLC26A2-related chondrodysplasias. FUND: This work was supported by National Natural Science Foundation of China (81871743, 81730065 and 81772377).
Publicações recentes
Computational biology insights into genotype-clinical phenotype-protein phenotype relationships between novel SLC26A2 variants identified in inherited skeletal dysplasias.
Suppressing UPR-dependent overactivation of FGFR3 signaling ameliorates SLC26A2-deficient chondrodysplasias.
A compound heterozygote SLC26A2 mutation resulting in robin sequence, mild limbs shortness, accelerated carpal ossification, and multiple epiphysial dysplasia in two Brazilian sisters. A new intermediate phenotype between diastrophic dysplasia and recessive multiple epiphyseal dysplasia.
Atelosteogenesis type I: autopsy findings.
Sulfate in fetal development.
📚 EuropePMC43 artigos no totalmostrando 2
Computational biology insights into genotype-clinical phenotype-protein phenotype relationships between novel SLC26A2 variants identified in inherited skeletal dysplasias.
European journal of medical geneticsSuppressing UPR-dependent overactivation of FGFR3 signaling ameliorates SLC26A2-deficient chondrodysplasias.
EBioMedicineAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Atelosteogênese II.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Atelosteogênese II
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Computational biology insights into genotype-clinical phenotype-protein phenotype relationships between novel SLC26A2 variants identified in inherited skeletal dysplasias.
- Suppressing UPR-dependent overactivation of FGFR3 signaling ameliorates SLC26A2-deficient chondrodysplasias.
- A compound heterozygote SLC26A2 mutation resulting in robin sequence, mild limbs shortness, accelerated carpal ossification, and multiple epiphysial dysplasia in two Brazilian sisters. A new intermediate phenotype between diastrophic dysplasia and recessive multiple epiphyseal dysplasia.
- Atelosteogenesis type I: autopsy findings.
- Sulfate in fetal development.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:56304(Orphanet)
- OMIM OMIM:256050(OMIM)
- MONDO:0009727(MONDO)
- GARD:8329(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q2870197(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Atelosteogênese II
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata