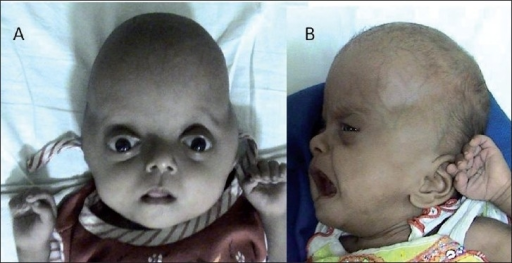
A síndrome de Pfeiffer tipo 1 (SP1) é uma forma da síndrome de Pfeiffer (SP) de gravidade leve a moderada. Ela se caracteriza pelo fechamento precoce das duas suturas coronais do crânio (as "costuras" que conectam os ossos da parte de cima da cabeça), por deformidades variadas nos dedos das mãos e dos pés e, na maioria dos casos, por um desenvolvimento intelectual normal.
Introdução
O que você precisa saber de cara
A síndrome de Pfeiffer tipo 1 (SP1) é uma forma da síndrome de Pfeiffer (SP) de gravidade leve a moderada. Ela se caracteriza pelo fechamento precoce das duas suturas coronais do crânio (as "costuras" que conectam os ossos da parte de cima da cabeça), por deformidades variadas nos dedos das mãos e dos pés e, na maioria dos casos, por um desenvolvimento intelectual normal.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 7 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 21 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Not applicable.
Curadoria gene-doença
fontes oficiaisTyrosine-protein kinase that acts as a cell-surface receptor for fibroblast growth factors and plays an essential role in the regulation of cell proliferation, differentiation, migration and apoptosis, and in the regulation of embryonic development. Required for normal embryonic patterning, trophoblast function, limb bud development, lung morphogenesis, osteogenesis and skin development. Plays an essential role in the regulation of osteoblast differentiation, proliferation and apoptosis, and is
Cell membraneGolgi apparatusCytoplasmic vesicleSecreted
Crouzon syndrome
An autosomal dominant syndrome characterized by craniosynostosis, hypertelorism, exophthalmos and external strabismus, parrot-beaked nose, short upper lip, hypoplastic maxilla, and a relative mandibular prognathism.
Tyrosine-protein kinase that acts as a cell-surface receptor for fibroblast growth factors and plays an essential role in the regulation of embryonic development, cell proliferation, differentiation and migration. Required for normal mesoderm patterning and correct axial organization during embryonic development, normal skeletogenesis and normal development of the gonadotropin-releasing hormone (GnRH) neuronal system. Phosphorylates PLCG1, FRS2, GAB1 and SHB. Ligand binding leads to the activati
Cell membraneNucleusCytoplasm, cytosolCytoplasmic vesicle
Pfeiffer syndrome
A syndrome characterized by the association of craniosynostosis, broad and deviated thumbs and big toes, and partial syndactyly of the fingers and toes. Three subtypes are known: mild autosomal dominant form (type 1); cloverleaf skull, elbow ankylosis, early death, sporadic (type 2); craniosynostosis, early demise, sporadic (type 3).
Variantes genéticas (ClinVar)
776 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 858 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
34 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Pfeiffer tipo 1
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Pfeiffer Syndrome (Acrocephalosyndactyly) With Significant Syndactyly and Brachydactyly: A Case Report.
We report a case of acrocephalosyndactyly, Pfeiffer syndrome type 1 with a mutation in FGFR2 c.758C>G (p.Ser253Trp) in a newborn with mild midfacial hypoplasia, significant brachydactyly and syndactyly in the hands and feet. One of the hallmark features of Pfeiffer syndrome is webbing or fusion (syndactyly) of the fingers and toes, which can vary in severity affecting both hands and feet. This variable expressivity of Pfeiffer syndrome makes classification of the condition challenging. Diagnosis was confirmed by genetic testing. Imaging investigations, clinical observation and physical examination further highlights the importance of interdisciplinary care involving orthopedic, neurosurgeons, geneticists, and pediatricians. Long-term follow-up is essential to monitor growth and development, while addressing associated complications including hearing loss and tracheal stenosis. This case underscores the complexity of acrocephalosyndactyly and its varying presentation. The baby was born at 35 weeks to non-consanguineous parents, with craniosynostosis, midfacial hypoplasia, broad thumbs, and toes.
Prevention and management of hearing loss in syndromic craniosynostosis: A case series.
To assess the audiological profile in a cohort of children affected by syndromic craniosynostosis. Eleven children with Apert syndrome (n=4), Saethre-Chotzen syndrome (n=3), Muenke syndrome (n=2), Crouzon syndrome (n=1) and Pfeiffer syndrome type 1 (n=1) were submitted to a complete audiologic evaluation including otoscopy, pure-tone audiometry, tympanometry and acoustic reflex testing, ABR, otoacustic emissions, temporal bone High Resolution CT (HRCT) scan. The main outcome measures were prevalence, type and severity of hearing loss, prevalence of chronic otitis media, correlation with the time of first surgical correction. Seven of 11 patients (64%) presented hearing loss (HL), conductive in 3/7 patients (43%) and mixed in 4/7 (57%). No patients showed a purely sensorineural HL. All hearing impaired patients displayed middle ear disorders: the patients with conductive HL had otitis media with effusion (OME) and 3/4 patients with mixed HL showed tympanic alterations or cholesteatoma. A bilateral vestibular aqueduct enlargement was detected by HRCT scan in one normal hearing patient. The ABRs resulted normal in all cases. Our study confirms the high prevalence of otologic diseases in such patients. In contrast with previous studies, middle ear disorders were responsible for the hearing impairment also in patients with mixed HL due to secondary inner ear damage. These findings restate the necessity of a close audiologic follow-up. We did not detect the specific ABR abnormalities previously reported, possibly because of an early correction of the cranial vault malformations.
Publicações recentes
Pfeiffer Syndrome (Acrocephalosyndactyly) With Significant Syndactyly and Brachydactyly: A Case Report.
Prevention and management of hearing loss in syndromic craniosynostosis: A case series.
Monoblock craniofacial internal distraction in a child with Pfeiffer syndrome: a case report.
A case of Pfeiffer syndrome type 1 with an A344P mutation in the FGFR2 gene.
📚 EuropePMC175 artigos no totalmostrando 2
Pfeiffer Syndrome (Acrocephalosyndactyly) With Significant Syndactyly and Brachydactyly: A Case Report.
Clinical medicine insights. Case reportsPrevention and management of hearing loss in syndromic craniosynostosis: A case series.
International journal of pediatric otorhinolaryngologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Pfeiffer tipo 1.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Pfeiffer tipo 1
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Pfeiffer Syndrome (Acrocephalosyndactyly) With Significant Syndactyly and Brachydactyly: A Case Report.
- Prevention and management of hearing loss in syndromic craniosynostosis: A case series.
- Monoblock craniofacial internal distraction in a child with Pfeiffer syndrome: a case report.
- A case of Pfeiffer syndrome type 1 with an A344P mutation in the FGFR2 gene.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:93258(Orphanet)
- MONDO:0019659(MONDO)
- GARD:16807(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55788781(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome Pfeiffer tipo 1
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata