A Síndrome de Pfeiffer tipo 2 (SP2) é um tipo comum e grave da Síndrome de Pfeiffer (SP), caracterizada por crânio em formato de trevo, problemas funcionais graves associados e alterações nas mãos, pés, cotovelos e joelhos.
Introdução
O que você precisa saber de cara
A Síndrome de Pfeiffer tipo 2 (SP2) é um tipo comum e grave da Síndrome de Pfeiffer (SP), caracterizada por crânio em formato de trevo, problemas funcionais graves associados e alterações nas mãos, pés, cotovelos e joelhos.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 10 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 35 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant, Not applicable.
Tyrosine-protein kinase that acts as a cell-surface receptor for fibroblast growth factors and plays an essential role in the regulation of cell proliferation, differentiation, migration and apoptosis, and in the regulation of embryonic development. Required for normal embryonic patterning, trophoblast function, limb bud development, lung morphogenesis, osteogenesis and skin development. Plays an essential role in the regulation of osteoblast differentiation, proliferation and apoptosis, and is
Cell membraneGolgi apparatusCytoplasmic vesicleSecreted
Crouzon syndrome
An autosomal dominant syndrome characterized by craniosynostosis, hypertelorism, exophthalmos and external strabismus, parrot-beaked nose, short upper lip, hypoplastic maxilla, and a relative mandibular prognathism.
Variantes genéticas (ClinVar)
308 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 858 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
17 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Pfeiffer tipo 2
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
2 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Prenatal diagnosis of Pfeiffer syndrome type 2 with increased nuchal translucency.
Pfeiffer syndrome (PS) is a rare autosomal dominant genetic disorder characterized by craniosynostosis, broad thumbs / toes. Here, we report a case of PS type 2 with increased nuchal translucency in early trimester.
Autopsy Case of Pfeiffer Syndrome Type 2, a Phenotype of Fibroblast Growth Factor Receptor-Associated Craniosynostosis Syndromes, with Tracheal Cartilage Sleeve and Abnormal Hyperplasia of Bronchial Cartilages.
BACKGROUND Pfeiffer syndrome (PS) is a fibroblast growth factor receptor (FGFR)-associated craniosynostosis syndrome, characterized by abnormally broad and medially deviated thumbs and great toes. Tracheal cartilage sleeve (TCS) is associated with several FGFR-associated craniosynostosis syndromes, including PS. TCS is an airway malformation in which the tracheal cartilage rings fuse with each other to form a sleeve of cartilage. CASE REPORT The patient was a 4-year-old girl with PS, TCS, and abnormal hyperplasia of non-fused intrapulmonary cartilages. The patient showed cranial dysplasia on prenatal ultrasonography. At birth, a cloverleaf skull in association with hydrocephalus and digital malformations was apparent. These findings were consistent with PS type 2. The diagnosis of PS type 2 was confirmed from a genetic test detecting a FGFR2 mutation (Y340C). During the clinical course, she underwent several surgeries, including ventriculoperitoneal shunts, sequential cranioplasty surgeries, and tracheotomy due to upper airway abnormalities. At 4 years old, she died of multiple organ failure following aspiration pneumonia. The autopsy revealed that the tracheal cartilages had fused with each other, resulting in a condition called TCS, in which the cartilage rings and tracheal ligaments were absent. The lungs were poorly aerated, and the dilated bronchi had thickened walls surrounded by many cartilage fragments, mainly at the hilum. These cartilages tended to overlap at both ends, did not fuse, and were greatly altered in size and shape. CONCLUSIONS We report the results of autopsy for PS with the first histopathological findings for the lungs and other visceral organs.
Pfeiffer Syndrome type 2; A case report of cranio-orbitofaciostenosis with bilateral choanal atresia at Muhimbili National Hospital, Tanzania.
Pfeiffer syndrome is a rare genetic disorder with heterogenous phenotype and prognosis. Due to its diverse clinical presentation, it can easily be misdiagnosed. Where genetic testing still remains a challenge, antenatal sonogram can aid in early diagnosis. The cranioorbito-faciostenosis demands aggressive management to permit survival instead of uniform early demise.
Pfeiffer type 2 syndrome: review with updates on its genetics and molecular biology.
Pfeiffer syndrome is a rare autosomal dominant inherited disorder associated with craniosynostosis, midfacial hypoplasia, and broad thumbs and toes. The syndrome has been divided into three clinical subtypes based on clinical findings. This review will specifically examine the most severe type, Pfeiffer syndrome type 2, focusing on its genetics and molecular biology. This subtype of the syndrome is caused by de novo sporadic mutations, the majority of which occur in the fibroblast growth factor receptor type 1 and 2 (FGFR1/2) genes. There is not one specific mutation, however. This disorder is genetically heterogeneous and may have varying phenotypic expressions that in various cases have overlapped with other similar craniosynostoses. A specific missense mutation of FGFR2 causing both Pfeiffer and Crouzon syndromes has been identified, with findings suggesting that gene expression may be affected by polymorphism within the same gene. Compared to other craniosynostosis-related disorders, Pfeiffer syndrome is the most extreme phenotype, as the underlying mutations cause wider effects on the secondary and tertiary protein structures and exhibit harsher clinical findings.
Pfeiffer type 2 syndrome: review with updates on its genetics and molecular biology.
Pfeiffer syndrome is a rare autosomal dominant inherited disorder associated with craniosynostosis, midfacial hypoplasia, and broad thumbs and toes. The syndrome has been divided into three clinical subtypes based on clinical findings. This review will specifically examine the most severe type, Pfeiffer syndrome type 2, focusing on its genetics and molecular biology. This subtype of the syndrome is caused by de novo sporadic mutations, the majority of which occur in the fibroblast growth factor receptor type 1 and 2 (FGFR1/2) genes. There is not one specific mutation, however. This disorder is genetically heterogeneous and may have varying phenotypic expressions that in various cases have overlapped with other similar craniosynostoses. A specific missense mutation of FGFR2 causing both Pfeiffer and Crouzon syndromes has been identified, with findings suggesting that gene expression may be affected by polymorphism within the same gene. Compared to other craniosynostosis-related disorders, Pfeiffer syndrome is the most extreme phenotype, as the underlying mutations cause wider effects on the secondary and tertiary protein structures and exhibit harsher clinical findings.
Publicações recentes
Prenatal diagnosis of Pfeiffer syndrome type 2 with increased nuchal translucency.
Autopsy Case of Pfeiffer Syndrome Type 2, a Phenotype of Fibroblast Growth Factor Receptor-Associated Craniosynostosis Syndromes, with Tracheal Cartilage Sleeve and Abnormal Hyperplasia of Bronchial Cartilages.
Pfeiffer Syndrome type 2; A case report of cranio-orbitofaciostenosis with bilateral choanal atresia at Muhimbili National Hospital, Tanzania.
Pfeiffer type 2 syndrome: review with updates on its genetics and molecular biology.
Pfeiffer type 2 syndrome: review with updates on its genetics and molecular biology.
📚 EuropePMC175 artigos no totalmostrando 6
Prenatal diagnosis of Pfeiffer syndrome type 2 with increased nuchal translucency.
Clinical case reportsAutopsy Case of Pfeiffer Syndrome Type 2, a Phenotype of Fibroblast Growth Factor Receptor-Associated Craniosynostosis Syndromes, with Tracheal Cartilage Sleeve and Abnormal Hyperplasia of Bronchial Cartilages.
The American journal of case reportsPfeiffer Syndrome type 2; A case report of cranio-orbitofaciostenosis with bilateral choanal atresia at Muhimbili National Hospital, Tanzania.
Clinical case reportsPfeiffer type 2 syndrome: review with updates on its genetics and molecular biology.
Child's nervous system : ChNS : official journal of the International Society for Pediatric NeurosurgeryMutation analysis of FGFR1-3 in 11 Japanese patients with syndromic craniosynostoses.
American journal of medical genetics. Part AA Delayed Finding of a Tracheal Cartilaginous Sleeve in a Patient with Pfeiffer Syndrome Type 2 and a Complex Airway History.
A & A case reportsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Pfeiffer tipo 2.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Pfeiffer tipo 2
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Prenatal diagnosis of Pfeiffer syndrome type 2 with increased nuchal translucency.
- Autopsy Case of Pfeiffer Syndrome Type 2, a Phenotype of Fibroblast Growth Factor Receptor-Associated Craniosynostosis Syndromes, with Tracheal Cartilage Sleeve and Abnormal Hyperplasia of Bronchial Cartilages.
- Pfeiffer Syndrome type 2; A case report of cranio-orbitofaciostenosis with bilateral choanal atresia at Muhimbili National Hospital, Tanzania.
- Pfeiffer type 2 syndrome: review with updates on its genetics and molecular biology.Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery· 2019· PMID 31222448mais citado
- Pfeiffer type 2 syndrome: review with updates on its genetics and molecular biology.Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery· 2019· PMID 30740633mais citado
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:93259(Orphanet)
- MONDO:0019660(MONDO)
- GARD:16808(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55788782(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
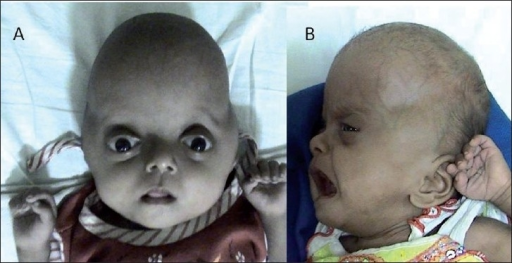
Síndrome Pfeiffer tipo 2
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata