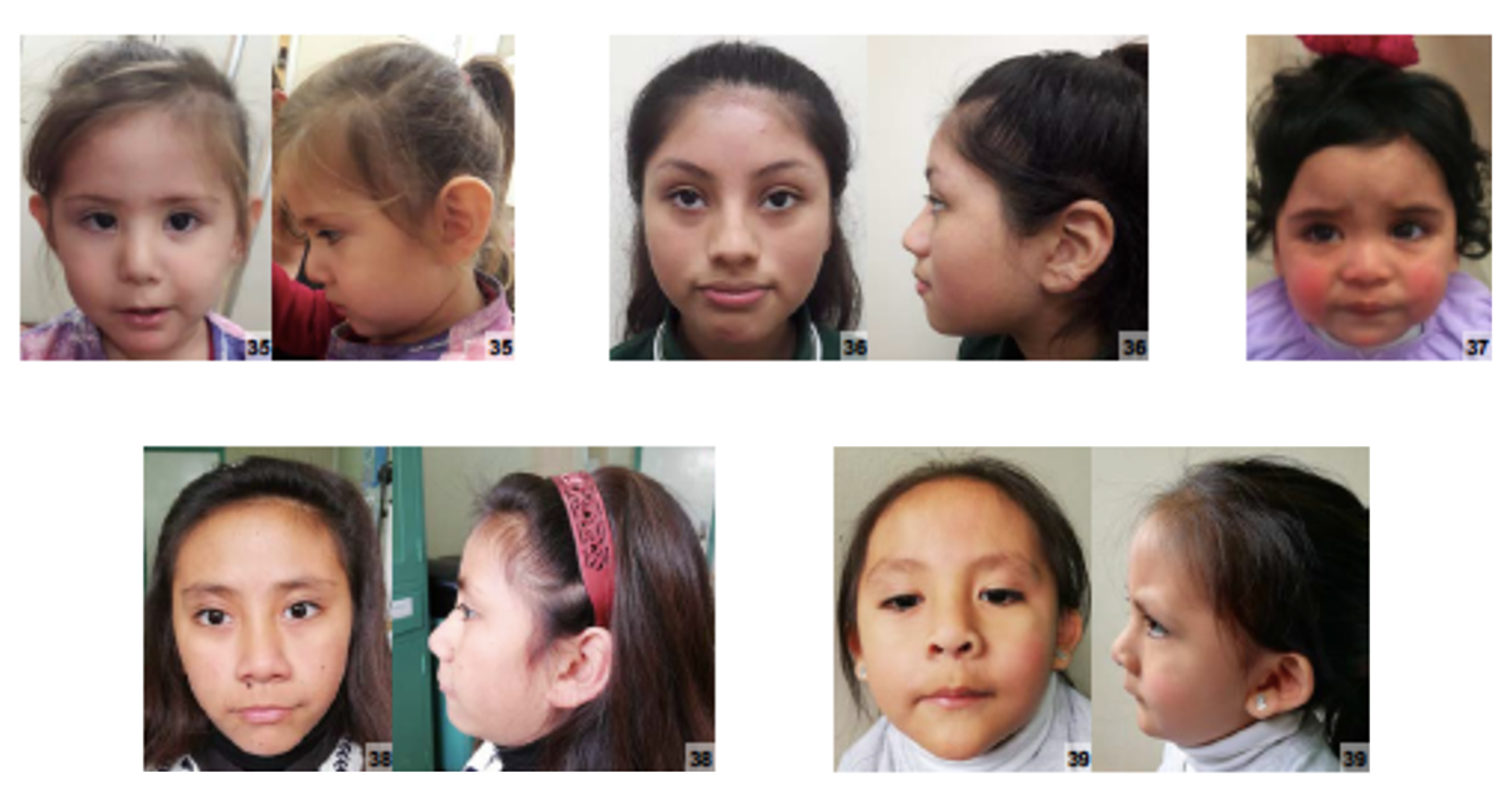
A monossomia distal 10q é uma anomalia cromossômica que envolve deleção terminal do braço longo do cromossomo 10 e é caracterizada por dismorfismo facial, retardo de crescimento pré e pós-natal, anomalias cardíacas e genitais e atraso no desenvolvimento.
Introdução
O que você precisa saber de cara
A monossomia distal 10q é uma anomalia cromossômica que envolve deleção terminal do braço longo do cromossomo 10 e é caracterizada por dismorfismo facial, retardo de crescimento pré e pós-natal, anomalias cardíacas e genitais e atraso no desenvolvimento.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 47 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 120 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Not applicable, Unknown.
Transcriptional activator (PubMed:28017370, PubMed:28017372, PubMed:28017373). Recognizes variations of the palindromic sequence 5'-ATTCCCNNGGGAATT-3' (By similarity)
Nucleus
Hypotonia, ataxia, and delayed development syndrome
An autosomal dominant neurodevelopmental syndrome characterized by global developmental delay, moderate to severe intellectual disability, cerebellar ataxia, hypotonia, speech delay, variable dysmorphic features, and genitourinary abnormalities including vesicoureteric reflux.
Variantes genéticas (ClinVar)
272 variantes patogênicas registradas no ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Deleção distal 10q
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Prenatal diagnosis of concomitant distal 5q duplication and terminal 10q deletion in a fetus with intrauterine growth restriction, congenital diaphragmatic hernia and congenital heart defects.
We present prenatal diagnosis of concomitant distal 5q duplication and terminal 10q deletion in a fetus with intrauterine growth restriction (IUGR), congenital diaphragmatic hernia (CDH) and congenital heart defects (CHD). A 34-year-old, gravida 4, para 2, woman was referred for amniocentesis at 21 weeks of gestation because of advanced maternal age and IUGR. There was no congenital malformation in the family. Amniocentesis revealed a derivative chromosome 10 with an additional maternal on the terminal region of 10q. Array comparative genomic hybridization (aCGH) analysis on the DNA extracted from the cultured amniocytes revealed a result of arr 5q31.3q35.5 (142, 548, 354-180,696,806) × 3.0, arr 10q26.3 (132, 932, 808-135,434,178) × 1.0 [GRCh37 (hg19)] with a 2.50-Mb deletion of 10q26.3 encompassing 19 [Online Mendelian Inheritance in Man (OMIM)] genes and a 38.15-Mb duplication of 5q31.3-q35.5 encompassing 195 OMIM genes including four CDH candidate genes of NDST1, ADAM19, NSD1 and MAML1. The mother was found to have a karyotype of 46,XX,t(5; 10) (q31.3; q26.3). Therefore, the fetal karyotype was 46,XX,der(10)t(5; 10)(q31.3; q26.3)mat. Prenatal ultrasound showed IUGR, right CDH, transposition of great artery, double outlet of right ventricle and right atrial isomerism. The pregnancy was terminated, and a malformed fetus was delivered with facial dysmorphism. Fetuses with concomitant distal 5q duplication and terminal 10q deletion may present IUGR, CDH and CHD on prenatal ultrasound.
A Patient with Trisomy 4p and Monosomy 10q.
Translocations are the most common structural abnormality in the human genome. Carriers of balanced chromosome rearrangements exhibit increased risk of abortion or a chromosomally-unbalanced child. The present study was carried out in 2017 at the Iranian Blood Transfusion Research Center. This study reported a rare chromosomal disorder with 4p duplication and 10q distal deletion syndrome which is associated with various complications at birth. Defects included the following characteristics: dysmorphic facial characteristic, hand or foot anomalies, growth retardation, developmental delay, strabismus, heart defects and renal anomalies. Cytogenetic analysis and array CGH were performed and, for the first time, we reported a patient with trisomy 4p16.3p12 and monosomy 10q26.3. The patient was found to have: arr 4p16.3p12 (37,152-45,490,207) x3, 10q26.3 (134,872,562-135,434,149) x1 genomic imbalances.
Expansion of the clinical phenotype of the distal 10q26.3 deletion syndrome to include ataxia and hyperemia of the hands and feet.
Distal deletion of the long arm of chromosome 10 is associated with a dysmorphic craniofacial appearance, microcephaly, behavioral issues, developmental delay, intellectual disability, and ocular, urogenital, and limb abnormalities. Herein, we present clinical, molecular, and cytogenetic investigations of four patients, including two siblings, with nearly identical terminal deletions of 10q26.3, all of whom have an atypical presentation of this syndrome. Their prominent features include ataxia, mild-to-moderate intellectual disability, and hyperemia of the hands and feet, and they do not display many of the other features commonly associated with deletions of this region. These results point to a novel gene locus associated with ataxia and highlight the variability of the clinical presentation of patients with deletions of this region.
10q26.1 Microdeletion: Redefining the critical regions for microcephaly and genital anomalies.
Distal 10q deletion syndrome is a well-characterized chromosomal disorder consisting of neurodevelopmental impairment, facial dysmorphism, cardiac malformations, genital and urinary tract defects, as well as digital anomalies. Patients with interstitial deletions involving band 10q26.1 present a phenotype similar to the ones with the distal 10q deletion syndrome, which led to the definition of a causal 600 kb smallest region of overlap (SRO). In this report, we describe a male patient with an interstitial 4.5 Mb deletion involving exclusively the 10q26.1 segment. He had growth and psychomotor retardation, microcephaly, flat feet, micropenis, and cryptorchidism. The patient's deleted region does not overlap the 10q SRO. We reviewed the clinical phenotype of patients with similar deletions and suggest the presence of two new SROs, one associated with microcephaly, growth and psychomotor retardation, and the other associated to genital anomalies. Interestingly, we narrowed those regions to segments encompassing five and two genes, respectively. FGFR2, NSMCE4A, and ATE1 were suggested as candidates for facial dysmorphism, growth cessation, and heart defects, respectively. WDR11 was linked to idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Its haploinsufficiency could play a crucial role in the genital anomalies of these patients.
Persistence of müllerian duct structures in a genetic male with distal monosomy 10q.
Persistent müllerian duct syndrome (PMD) with antimüllerian hormone (AMH) deficiency is usually associated with mutations or deletions of the AMH gene, although many cases have no identified gene association. We report on a genetic male with PMD and AMH deficiency associated with distal monosomy 10q. A term 3,230 g infant was born to a healthy 27-year-old. Fetal ultrasound had shown possible genital ambiguity. Postnatal exam showed a 0.5 cm phallus with basal meatus, normal scrotum with no palpable gonads, no vaginal orifice, and a rectal fistula with an imperforate anus. Voiding cystourethrogram with ultrasound, cystoscopy, and laparoscopy showed normal bladder, urethral orifice, distal vagina, cervix, and bilateral abdominal testis. At 24 hours of life, testosterone was within normal range with low AMH level. Chromosome microarray analysis showed 46, XY, del10(10q25.3q26.13) involving an 8.2 MB interstitial deletion. Whole exome sequencing identified a NOTCH2 variant (1p11.2). AMH sequencing revealed no abnormalities. Following multidisciplinary team and parent discussion, male gender was assigned. Testosterone treatment resulted in penile length of 1.5 cm. Bilateral orchiopexy and posterior sagittal anorectoplasty were performed at 11 months of age; rudimentary müllerian structures were identified. This observation suggests an association of 10qter elements with male differentiation including AMH expression and is similar to a patient with 46, XY, del(10q26.1) in which AMH levels were not reported. Regional candidate genes include FGFR2 (10q26.13). The possible contribution of a NOTCH2 variant cannot be excluded.
Publicações recentes
Corrigendum Re: "Persistence of müllerian duct structures in a genetic male with distal monosomy 10q. Am J Med Genet A. 2015 Apr;167A(4):791-6. Doi:10.1002/ajmg.a.37014".
Persistence of müllerian duct structures in a genetic male with distal monosomy 10q.
Familial complex 3q;10q rearrangement unraveled by subtelomeric FISH analysis.
📚 EuropePMCmostrando 5
Prenatal diagnosis of concomitant distal 5q duplication and terminal 10q deletion in a fetus with intrauterine growth restriction, congenital diaphragmatic hernia and congenital heart defects.
Taiwanese journal of obstetrics & gynecologyA Patient with Trisomy 4p and Monosomy 10q.
Archives of Iranian medicineExpansion of the clinical phenotype of the distal 10q26.3 deletion syndrome to include ataxia and hyperemia of the hands and feet.
American journal of medical genetics. Part A10q26.1 Microdeletion: Redefining the critical regions for microcephaly and genital anomalies.
American journal of medical genetics. Part APersistence of müllerian duct structures in a genetic male with distal monosomy 10q.
American journal of medical genetics. Part AAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Deleção distal 10q.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Deleção distal 10q
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Prenatal diagnosis of concomitant distal 5q duplication and terminal 10q deletion in a fetus with intrauterine growth restriction, congenital diaphragmatic hernia and congenital heart defects.
- A Patient with Trisomy 4p and Monosomy 10q.
- Expansion of the clinical phenotype of the distal 10q26.3 deletion syndrome to include ataxia and hyperemia of the hands and feet.
- 10q26.1 Microdeletion: Redefining the critical regions for microcephaly and genital anomalies.
- Persistence of müllerian duct structures in a genetic male with distal monosomy 10q.
- Corrigendum Re: "Persistence of müllerian duct structures in a genetic male with distal monosomy 10q. Am J Med Genet A. 2015 Apr;167A(4):791-6. Doi:10.1002/ajmg.a.37014".
- Familial complex 3q;10q rearrangement unraveled by subtelomeric FISH analysis.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:96148(Orphanet)
- OMIM OMIM:609625(OMIM)
- MONDO:0012315(MONDO)
- GARD:3711(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q21154055(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Deleção distal 10q
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata