A síndrome 48,XXXY é uma anomalia cromossômica que envolve um número anormal de cromossomos, caracterizada pela presença de dois cromossomos X extras em homens.
Introdução
O que você precisa saber de cara
A síndrome 48,XXXY é uma anomalia cromossômica que envolve um número anormal de cromossomos, caracterizada pela presença de dois cromossomos X extras em homens.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 20 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 62 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome 48,XXXY
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Incidence of miscarriages in women with children with 47,XXY, 48,XXXY, or 49,XXXXY.
The management and the causes of miscarriages present challenges for the obstetrical community as well as for families. In families who have a child with an X and Y chromosomal disorder, the duress of pregnancy loss may be significantly exacerbated. A greater understanding of these mechanisms would inform medical providers and families of children with 47,XXY, or Klinefelter syndrome (KS), and variant disorders (48,XXXY and 49,XXXXY). This study investigates miscarriage incidences across a cohort of mothers of male offspring born with these disorders. Pregnancy history of mothers of male offspring with 47,XXY (KS) or variant disorders was collected. Statistical analyses were performed to determine if these mothers experienced higher incidences of miscarriage than the general population. Mothers reporting miscarriage in the 47,XXY (p = 0.03, d = 0.31) and 48,XXXY (p = 0.02, d = 1.95) groups were significantly older at the time of birth than those who did not report miscarriage. When compared to known statistics of miscarriage in the general population, there was a significant increase in the miscarriages in the 47,XXY (p = 0.04, h = 0.13), 49,XXXXY (p = 0.02, h = 0.23), and combined groups (p < 0.01, h = 0.15). Mothers of children with 47,XXY (KS) and variant disorders are at increased risk of miscarriage compared to the general population based on the findings of this study.
From Klinefelter Syndrome to High Grade Aneuploidies: Expanding the Gene-dosage Effect of Supernumerary X Chromosomes.
High-grade aneuploidies of X and Y sex chromosomes (HGAs) are exceedingly rare and complex conditions. We aimed to investigate the effect of supernumerary X chromosomes (extra-Xs) on the clinical, hormonal, metabolic, and echocardiographic features of patients with HGAs. In a cross-sectional study, we compared 23 subjects with HGAs and 46 age-matched subjects with 47,XXY Klinefelter syndrome (KS), according to the number of extra-Xs: two (47,XXY and 48,XXYY), three (48,XXXY and 49,XXXYY), or four supernumerary Xs (49,XXXXY). A second cohort consisting of 46 pubertal stage-matched KS subjects was employed for validation. Clinical, hormonal, metabolic and ultrasonographic parameters were collected and analyzed. The increase in the number of extra-Xs was associated with a progressive adverse effect on height, pubertal development, testicular volume and function, adrenal steroidogenesis, and thyroid function. A progressive linear increase in ACTH and a decrease in cortisol/ACTH ratios were found. Weight and body mass index, Sertoli cell function, lipid profile, and glucose tolerance post-oral glucose tolerance test were all worse in the HGA cohort compared to KS. Cardiac evaluation revealed a linear association with reduced left and right end-diastolic diameters and reduced ejection fraction. The increase in the number of extra-Xs is associated with a "dose-dependent" progressive impairment in steroid producing glands, thyroid function, cardiac structure, and performance.
Generation of two iPSC lines (KAUSTi001-A, KAUSTi002-A) from a rare high-grade Klinefelter Syndrome patient (49-XXXXY) carrying a balanced translocation t(4,11) (q35,q23).
Klinefelter Syndrome (KS) is the most common aneuploidy in humans (prevalence: 85-250 per 100,000 born males) and is characterized by one or more supernumerary X-chromosomes (47-XXY, 48-XXXY and 49-XXXXY karyotypes). KS is a multisystemic disorder associated to multiple phenotypic features including cardiac abnormalities, infertility, mental retardation, diabetes and increased cancer risk. Using a non-integrative mRNAs reprogramming approach, we generated two iPSC lines 48-XXXY and 49-XXXXY from a non-mosaic 49-XXXXY KS patient carrying a balanced translocation t(4,11) (q35,q23). These iPSC lines provide a unique cellular platform to study the molecular mechanisms underlying KS pathophysiology.
Establishment of an iPSC cohort from three unrelated 47-XXY Klinefelter Syndrome patients (KAUSTi007-A, KAUSTi007-B, KAUSTi009-A, KAUSTi009-B, KAUSTi010-A, KAUSTi010-B).
Klinefelter Syndrome (KS) is caused by the presence of a supernumerary X chromosome. Cytogenetic studies revaled that 80-90% of patients carry a 47-XXY karyotype, while 10-20% of cases are represented by mosaic 46-XY/47-XXY and high-grade aneuploidies 48-XXXY and 48-XXYY. The phenotypic traits of KS are highly variable across individuals and include cognitive dysfunction, metabolic dysregulation, osteoporosis, and cardiovascular diseases. Here, we describe the derivation of multiple 47-XXY iPSC lines from three unrelated KS patients to study the impact of supernumerary X chromosome during early development.
Establishment of iPSC lines from a high-grade Klinefelter Syndrome patient (49-XXXXY) and two genetically matched healthy relatives (KAUSTi003-A, KAUSTi004-A, KAUSTi004-B, KAUSTi005-A, KAUSTi005-B, KAUSTi005-C).
Klinefelter Syndrome (KS) is the most frequent X chromosome aneuploidy in males. KS patients with 47-XXY, 48-XXXY and 49-XXXXY karyotypes endure inter-individual phenotypic variabilities including infertility, cardiac diseases, metabolic and psychiatric disorders. We derived iPSC lines from a high-grade 49-XXXXY KS and two healthy donors' fibroblasts. Importantly, the healthy controls XY and XX are direct relatives to KS patients, thus enabling functional comparisons of healthy and disease iPSCs with partially matched genetic backgrounds. These iPSC lines provide an unprecedented cellular tool to study KS pathophysiology at the pluripotent stage as well as during differentiation into disease relevant cell types.
Publicações recentes
Genetics, Cytogenetic Testing and Conventional Karyotype.
Incidence of miscarriages in women with children with 47,XXY, 48,XXXY, or 49,XXXXY.
Generating Advancements in Longitudinal Analysis in X and Y Variations: Rationale, Methods, and Diagnostic Characteristics for the GALAXY Registry.
Prevalence, spermatozoa, hormonal, and genetic evaluation of rare mosaic klinefelter syndrome patients in southern China.
A Mosaic PHEX Variant in Hypophosphatemic Rickets: Distinguishing Postzygotic Mutation from Sex Chromosome Aneuploidy.
📚 EuropePMCmostrando 5
Incidence of miscarriages in women with children with 47,XXY, 48,XXXY, or 49,XXXXY.
Frontiers in endocrinologyFrom Klinefelter Syndrome to High Grade Aneuploidies: Expanding the Gene-dosage Effect of Supernumerary X Chromosomes.
The Journal of clinical endocrinology and metabolismGeneration of two iPSC lines (KAUSTi001-A, KAUSTi002-A) from a rare high-grade Klinefelter Syndrome patient (49-XXXXY) carrying a balanced translocation t(4,11) (q35,q23).
Stem cell researchEstablishment of an iPSC cohort from three unrelated 47-XXY Klinefelter Syndrome patients (KAUSTi007-A, KAUSTi007-B, KAUSTi009-A, KAUSTi009-B, KAUSTi010-A, KAUSTi010-B).
Stem cell researchEstablishment of iPSC lines from a high-grade Klinefelter Syndrome patient (49-XXXXY) and two genetically matched healthy relatives (KAUSTi003-A, KAUSTi004-A, KAUSTi004-B, KAUSTi005-A, KAUSTi005-B, KAUSTi005-C).
Stem cell researchAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome 48,XXXY.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome 48,XXXY
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Incidence of miscarriages in women with children with 47,XXY, 48,XXXY, or 49,XXXXY.
- From Klinefelter Syndrome to High Grade Aneuploidies: Expanding the Gene-dosage Effect of Supernumerary X Chromosomes.
- Generation of two iPSC lines (KAUSTi001-A, KAUSTi002-A) from a rare high-grade Klinefelter Syndrome patient (49-XXXXY) carrying a balanced translocation t(4,11) (q35,q23).
- Establishment of an iPSC cohort from three unrelated 47-XXY Klinefelter Syndrome patients (KAUSTi007-A, KAUSTi007-B, KAUSTi009-A, KAUSTi009-B, KAUSTi010-A, KAUSTi010-B).
- Establishment of iPSC lines from a high-grade Klinefelter Syndrome patient (49-XXXXY) and two genetically matched healthy relatives (KAUSTi003-A, KAUSTi004-A, KAUSTi004-B, KAUSTi005-A, KAUSTi005-B, KAUSTi005-C).
- Genetics, Cytogenetic Testing and Conventional Karyotype.
- Generating Advancements in Longitudinal Analysis in X and Y Variations: Rationale, Methods, and Diagnostic Characteristics for the GALAXY Registry.
- Prevalence, spermatozoa, hormonal, and genetic evaluation of rare mosaic klinefelter syndrome patients in southern China.
- A Mosaic PHEX Variant in Hypophosphatemic Rickets: Distinguishing Postzygotic Mutation from Sex Chromosome Aneuploidy.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:96263(Orphanet)
- MONDO:0019928(MONDO)
- GARD:5676(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q8042637(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
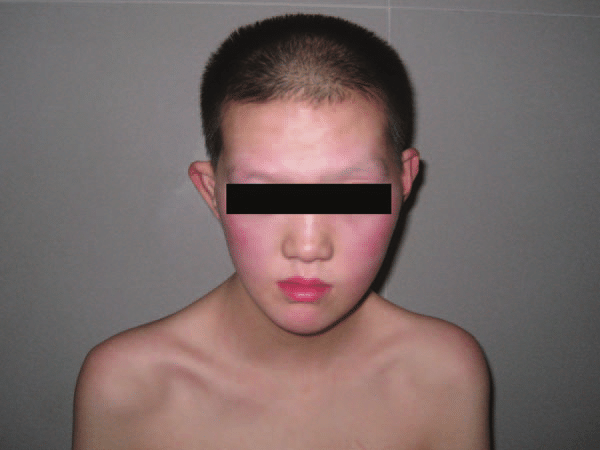
Síndrome 48,XXXY
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata