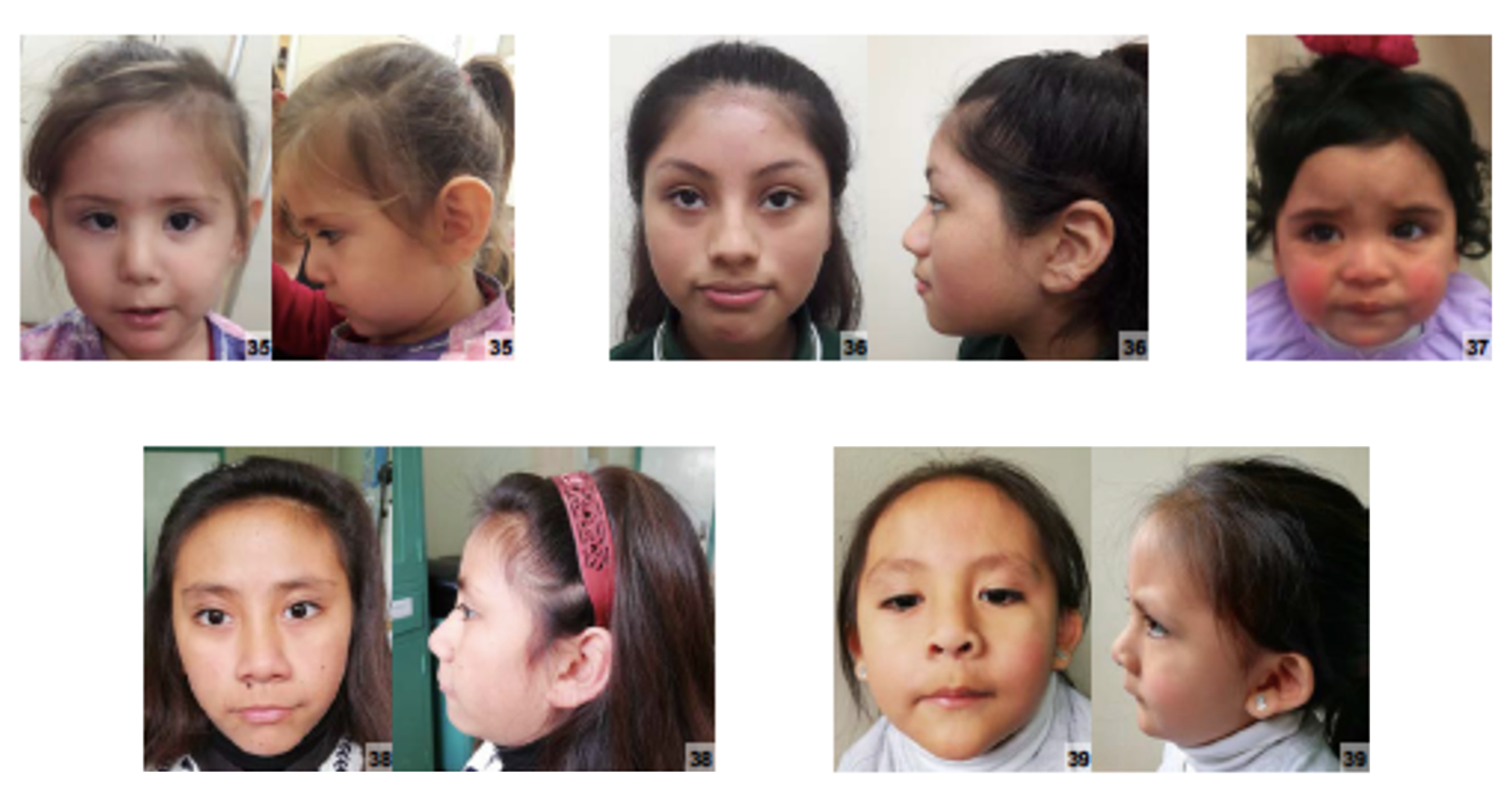
É uma condição genética ligada ao cromossomo X que causa deficiência intelectual e outros sintomas. Caracteriza-se por uma deficiência intelectual de moderada a grave em meninos e moderada em meninas. Foi observada em 14 pessoas de quatro gerações de uma mesma família. Foi relatado aumento do tamanho da cabeça (macrocefalia) e, em alguns casos, também pode haver uma malformação grave no cérebro (holoprosencefalia), como observado em duas pessoas da família. A forma de herança é genética, ligada ao cromossomo X e é semi-dominante.
Introdução
O que você precisa saber de cara
É uma condição genética ligada ao cromossomo X que causa deficiência intelectual e outros sintomas. Caracteriza-se por uma deficiência intelectual de moderada a grave em meninos e moderada em meninas. Foi observada em 14 pessoas de quatro gerações de uma mesma família. Foi relatado aumento do tamanho da cabeça (macrocefalia) e, em alguns casos, também pode haver uma malformação grave no cérebro (holoprosencefalia), como observado em duas pessoas da família. A forma de herança é genética, ligada ao cromossomo X e é semi-dominante.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 26 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 77 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição.
Curadoria gene-doença
fontes oficiaisE3 ubiquitin-protein ligase which mediates ubiquitination and subsequent proteasomal degradation of target proteins (PubMed:15567145, PubMed:15767685, PubMed:15989957, PubMed:17567951, PubMed:18488021, PubMed:19037095, PubMed:19713937, PubMed:20534529, PubMed:30217973). Regulates apoptosis by catalyzing the polyubiquitination and degradation of MCL1 (PubMed:15989957). Mediates monoubiquitination of DNA polymerase beta (POLB) at 'Lys-41', 'Lys-61' and 'Lys-81', thereby playing a role in base-exci
CytoplasmNucleusMitochondrion
Intellectual developmental disorder, X-linked, syndromic, Turner type
An X-linked neurodevelopmental disorder with highly variable clinical manifestations. Common features consist of moderate to profound intellectual disability, delayed or absent speech, short stature with small hands and feet, and non-specific but recurrent dysmorphic facial features such as macrocephaly, microcephaly, a broad nasal tip, deep set eyes, epicanthic folds, short palpebral fissures and a short philtrum. Patients may manifest other features, such as hypotonia, seizures and delayed bone age.
Variantes genéticas (ClinVar)
698 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Perturbação do desenvolvimento intelectual ligada ao X, tipo Turner
Centros de Referência SUS
37 centros habilitados pelo SUS para Perturbação do desenvolvimento intelectual ligada ao X, tipo Turner
Centros para Perturbação do desenvolvimento intelectual ligada ao X, tipo Turner
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
An International Longitudinal Natural History Study of Patients With Danon Disease: Unique Cardiac Trajectories Identified Based on Sex and Heart Failure Outcomes.
Danon disease is a rare X-linked dominant cardioskeletal myopathy caused by mutations in the lysosome-associated membrane protein-2 gene. Though the severe morbidity of disease in men is well established, longitudinal studies describing the trajectory of cardiovascular disease in both sexes are lacking. Data were from the International Danon Disease Registry and includes retrospective data on 116 patients who provided informed consent between 2005 and 2022. The analysis included 116 patients with Danon disease with a median age of diagnosis of 15.2 years (10.0-25.2 years). Trends in echocardiographic parameters over time indicate decreased left ventricular ejection fraction and increased left ventricular end diastolic dimension regardless of sex and heart failure (HF) outcome; however, rate of change was increased in patients who experienced a HF outcome in both sexes. Left ventricular wall hypertrophy continues in men who have not yet experienced a HF outcome and stabilizes before HF outcome, while females have progressive left ventricular thinning regardless of HF outcome. Stratified analysis demonstrated 2 distinct groups of females: one who experienced HF outcome before 26 years of age and another group with this HF outcome after. In this largest longitudinal natural history study of Danon disease to date, we confirmed that males present on average a decade earlier and demonstrate more progressive cardiac hypertrophy and HF than females. Notably, there may be a subset of females who are phenotypically similar to males with profound left ventricular hypertrophy that appears to stabilize or regress before HF outcome. Correlations between structural cardiac dysfunction and disease progression may permit risk stratification, refinement of treatment algorithms, and inform therapeutic trial design. URL: https://clinicaltrials.gov/; Unique Identifier: NCT03766386.
Exploring the Clinical Spectrum of HUWE1 -Related Neurodevelopmental Disorder: Five New Patients and Literature Review.
Turner-type X-linked syndromic intellectual developmental disorder (MRXST) is a neurodevelopmental disorder associated with variants in the HUWE1 gene on chromosome Xp11. The condition is characterized by variable phenotypes, including global developmental delay, intellectual disability, and distinctive facial dysmorphisms, with inheritance patterns ranging from X-linked recessive to de novo mutations in females. Here, we describe five probands in two families, highlighting their clinical features and genetic findings. Trio whole-exome sequencing identified a de novo variant in HUWE1 in the proband in one family and a maternally inherited hemizygous variant in three boys in a second family. A comprehensive review of HUWE1-associated cases from the literature assisted genotype-phenotype correlations, revealing consistent features such as intellectual disability, skeletal anomalies, and facial dysmorphisms as well as instances of intrafamilial variability. Our findings confirm the phenotypic variability of MRXST and underscore the significance of the HUWE1 gene product in neurodevelopment. We propose a baseline monitoring protocol to aid in diagnosis and management, contributing to the development of specific guidelines for patient follow-up.
Calcium-Dependent Regulation of Neuronal Excitability Is Rescued in Fragile X Syndrome by a Tat-Conjugated N-Terminal Fragment of FMRP.
Fragile X syndrome (FXS) arises from the loss of fragile X messenger ribonucleoprotein (FMRP) needed for normal neuronal excitability and circuit functions. Recent work revealed that FMRP contributes to mossy fiber long-term potentiation by adjusting the Kv4 A-type current availability through interactions with a Cav3-Kv4 ion channel complex, yet the mechanism has not yet been defined. In this study using wild-type and Fmr1 knock-out (KO) tsA-201 cells and cerebellar sections from male Fmr1 KO mice, we show that FMRP associates with all subunits of the Cav3.1-Kv4.3-KChIP3 complex and is critical to enabling calcium-dependent shifts in Kv4.3 inactivation to modulate the A-type current. Specifically, upon depolarization Cav3 calcium influx activates dual-specific phosphatase 1/6 (DUSP1/6) to deactivate ERK1/2 (ERK) and lower phosphorylation of Kv4.3, a signaling pathway that does not function in Fmr1 KO cells. In Fmr1 KO mouse tissue slices, cerebellar granule cells exhibit a hyperexcitable response to membrane depolarizations. Either incubating Fmr1 KO cells or in vivo administration of a tat-conjugated FMRP N-terminus fragment (FMRP-N-tat) rescued Cav3-Kv4 function and granule cell excitability, with a decrease in the level of DUSP6. Together these data reveal a Cav3-activated DUSP signaling pathway critical to the function of a FMRP-Cav3-Kv4 complex that is misregulated in Fmr1 KO conditions. Moreover, FMRP-N-tat restores function of this complex to rescue calcium-dependent control of neuronal excitability as a potential therapeutic approach to alleviating the symptoms of FXS.
Facial and ocular manifestations of male patients affected by the HUWE1-related intellectual developmental disorder.
Turner-type X-linked syndromic intellectual developmental disorder (MRXST) is a rare neurodevelopmental disorder. MRXST is caused by pathogenic variants in the HUWE1 gene on chromosome Xp11.22. The HUWE1 gene encodes a ubiquitin ligase, which has downstream effects on the n-MYC protein and DLL3 Notch ligand, ultimately affecting neuronal differentiation. In addition to intellectual disability and developmental delay, other clinical features such as absent or delayed speech, skeletal abnormalities, abnormalities in hands or feet, seizures, and hypotonia have been described in case reports. Facial dysmorphic features and eye manifestations have been reported in patients with MRXST, but have not been identified as distinctive to this condition. We report two cases of individuals affected by HUWE1-Related Intellectual Developmental Disorder and present a review of literature of male patients affected by this disorder. Based on the literature review and findings in our two patients, it is our observation that patients with MRXST present with distinctive features, which include broad nasal tip, root, or prominent nose (39%), blepharophimosis (27%), epicanthic folds (25%), ear abnormalities (25%), thin upper lip (23%), and deep set eyes (23%). Furthermore, we note that oculofacial abnormalities are seen more frequently in patients with missense variants than patients with duplications in the HUWE1 gene. The findings noted in this paper may help clinicians suspect a diagnosis of MRXST when presented with these distinctive ocular and facial features.
FMRP(1-297)-tat restores ion channel and synaptic function in a model of Fragile X syndrome.
Fragile X Syndrome results from a loss of Fragile X Mental Retardation Protein (FMRP). We now show that FMRP is a member of a Cav3-Kv4 ion channel complex that is known to regulate A-type potassium current in cerebellar granule cells to produce mossy fiber LTP. Mossy fiber LTP is absent in Fmr1 knockout (KO) mice but is restored by FMRP(1-297)-tat peptide. This peptide further rapidly permeates the blood-brain barrier to enter cells across the cerebellar-cortical axis that restores the balance of protein translation for at least 24 h and transiently reduces elevated levels of activity of adult Fmr1 KO mice in the Open Field Test. These data reveal that FMRP(1-297)-tat can improve function from the levels of protein translation to synaptic efficacy and behaviour in a model of Fragile X syndrome, identifying a potential therapeutic strategy for this genetic disorder.
Publicações recentes
Renpenning syndrome caused by the c.459_462delAGAG mutation in PQBP1: a case report and literature review.
35 Individuals With HUWE1-Related Neurodevelopmental Disorder and Suggested Clinical Evaluations.
ARHGEF6-dependent cytoskeletal regulation underlies a conserved program of forebrain interneuron development.
A Novel MID1 Mutation Identified in a Patient With Craniofacial Anomalies and X-Linked Intellectual Disability.
Emerging role of KDM5C in X-linked intellectual disability based on human genetic data and zebrafish models.
📚 EuropePMCmostrando 9
An International Longitudinal Natural History Study of Patients With Danon Disease: Unique Cardiac Trajectories Identified Based on Sex and Heart Failure Outcomes.
Journal of the American Heart AssociationExploring the Clinical Spectrum of HUWE1 -Related Neurodevelopmental Disorder: Five New Patients and Literature Review.
American journal of medical genetics. Part ACalcium-Dependent Regulation of Neuronal Excitability Is Rescued in Fragile X Syndrome by a Tat-Conjugated N-Terminal Fragment of FMRP.
The Journal of neuroscience : the official journal of the Society for NeuroscienceFacial and ocular manifestations of male patients affected by the HUWE1-related intellectual developmental disorder.
International journal of molecular epidemiology and geneticsFMRP(1-297)-tat restores ion channel and synaptic function in a model of Fragile X syndrome.
Nature communicationsCase report: a rare case of Hunter syndrome (type II mucopolysaccharidosis) in a girl.
BMC medical geneticsCREB Signaling Is Involved in Rett Syndrome Pathogenesis.
The Journal of neuroscience : the official journal of the Society for NeuroscienceMutation Update for Kabuki Syndrome Genes KMT2D and KDM6A and Further Delineation of X-Linked Kabuki Syndrome Subtype 2.
Human mutationHUWE1 mutations in Juberg-Marsidi and Brooks syndromes: the results of an X-chromosome exome sequencing study.
BMJ openAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Perturbação do desenvolvimento intelectual ligada ao X, tipo Turner.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Perturbação do desenvolvimento intelectual ligada ao X, tipo Turner
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- An International Longitudinal Natural History Study of Patients With Danon Disease: Unique Cardiac Trajectories Identified Based on Sex and Heart Failure Outcomes.
- Exploring the Clinical Spectrum of HUWE1 -Related Neurodevelopmental Disorder: Five New Patients and Literature Review.
- Calcium-Dependent Regulation of Neuronal Excitability Is Rescued in Fragile X Syndrome by a Tat-Conjugated N-Terminal Fragment of FMRP.The Journal of neuroscience : the official journal of the Society for Neuroscience· 2024· PMID 38664011mais citado
- Facial and ocular manifestations of male patients affected by the HUWE1-related intellectual developmental disorder.
- FMRP(1-297)-tat restores ion channel and synaptic function in a model of Fragile X syndrome.
- Renpenning syndrome caused by the c.459_462delAGAG mutation in PQBP1: a case report and literature review.
- 35 Individuals With HUWE1-Related Neurodevelopmental Disorder and Suggested Clinical Evaluations.
- ARHGEF6-dependent cytoskeletal regulation underlies a conserved program of forebrain interneuron development.
- A Novel MID1 Mutation Identified in a Patient With Craniofacial Anomalies and X-Linked Intellectual Disability.
- Emerging role of KDM5C in X-linked intellectual disability based on human genetic data and zebrafish models.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:85328(Orphanet)
- OMIM OMIM:300612(OMIM)
- MONDO:0010407(MONDO)
- GARD:81(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q28065615(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Perturbação do desenvolvimento intelectual ligada ao X, tipo Turner
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata