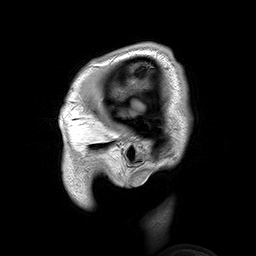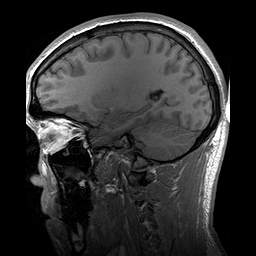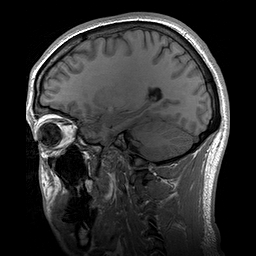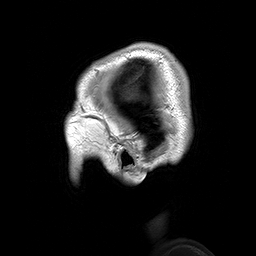
A Osteopatia Estriada com Esclerose Craniana (OS-CS) é uma displasia óssea, ou seja, uma condição que causa problemas no desenvolvimento dos ossos. Ela é caracterizada por linhas longitudinais nas metáfises (as partes dos ossos longos onde eles crescem), endurecimento dos ossos do crânio e do rosto, macrocefalia (cabeça maior que o normal), fenda palatina (o conhecido "céu da boca rachado") e perda de audição.
Introdução
O que você precisa saber de cara
A Osteopatia Estriada com Esclerose Craniana (OS-CS) é uma displasia óssea, ou seja, uma condição que causa problemas no desenvolvimento dos ossos. Ela é caracterizada por linhas longitudinais nas metáfises (as partes dos ossos longos onde eles crescem), endurecimento dos ossos do crânio e do rosto, macrocefalia (cabeça maior que o normal), fenda palatina (o conhecido "céu da boca rachado") e perda de audição.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 31 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 100 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: X-linked dominant.
Curadoria gene-doença
fontes oficiaisKey downstream component of the canonical Wnt signaling pathway (PubMed:17524503, PubMed:18077326, PubMed:18086858, PubMed:18957423, PubMed:21262353, PubMed:22155184, PubMed:22647378, PubMed:22699938). In the absence of Wnt, forms a complex with AXIN1, AXIN2, APC, CSNK1A1 and GSK3B that promotes phosphorylation on N-terminal Ser and Thr residues and ubiquitination of CTNNB1 via BTRC and its subsequent degradation by the proteasome (PubMed:17524503, PubMed:18077326, PubMed:18086858, PubMed:189574
CytoplasmNucleusCytoplasm, cytoskeletonCell junction, adherens junctionCell junctionCell membraneCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, spindle poleSynapseCytoplasm, cytoskeleton, cilium basal body
Colorectal cancer
A complex disease characterized by malignant lesions arising from the inner wall of the large intestine (the colon) and the rectum. Genetic alterations are often associated with progression from premalignant lesion (adenoma) to invasive adenocarcinoma. Risk factors for cancer of the colon and rectum include colon polyps, long-standing ulcerative colitis, and genetic family history.
Regulator of the canonical Wnt signaling pathway. Acts by specifically binding phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2), translocating to the cell membrane and interacting with key regulators of the canonical Wnt signaling pathway, such as components of the beta-catenin destruction complex. Acts both as a positive and negative regulator of the Wnt signaling pathway, depending on the context: acts as a positive regulator by promoting LRP6 phosphorylation. Also acts as a negative regu
CytoplasmCell membraneNucleus
Osteopathia striata with cranial sclerosis
An X-linked dominant sclerosing bone dysplasia that presents in females with macrocephaly, cleft palate, facial palsy, conductive hearing loss, mild learning disabilities, sclerosis of the long bones and skull. Longitudinal striations are visible on radiographs of the long bones, pelvis, and scapulae (osteopathia striata). In males this entity is usually associated with fetal or neonatal lethality. Occasional surviving males have, in addition to hyperostosis, cardiac, intestinal, and genitourinary malformations.
Variantes genéticas (ClinVar)
584 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
35 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de osteopatia estriada-esclerose craniana
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Mostrando amostra de 4 publicações de um total de 40
Osteopathia striata with cranial sclerosis as a cancer predisposition syndrome: The first report of neuroblastoma and review of all cancers in OSCS.
Osteopathia Striata with Cranial Sclerosis (OSCS) is a rare genetic condition primarily characterized by metaphyseal striations of long bones, bone sclerosis, macrocephaly, and other congenital anomalies. It is caused by pathogenic variants in AMER1, a tumor suppressor and a WNT signaling repressor gene with key roles in tissue regeneration, neurodevelopment, tumorigenesis, and other developmental processes. While somatic AMER1 pathogenic variants have frequently been identified in several tumor types (e.g., Wilms tumor and colorectal cancer), whether OSCS (i.e., with AMER1 germline variants) is a tumor predisposition syndrome is not clear, with only nine cases reported with tumors. We here report the first case of neuroblastoma diagnosed in a male child with OSCS, review all previously reported tumors diagnosed in individuals with OSCS, and discuss potential tumorigenic mechanisms of AMER1. Our report adds to the accumulating evidence suggesting OSCS is a tumor predisposition condition, highlighting the importance of maintaining a high index of suspicion for the associated tumors when evaluating patients with OSCS. Importantly, Wilms tumor stands out as the most commonly observed tumor in OSCS patients, underscoring the need for regular surveillance.
Intestinal malrotation in a female newborn affected by Osteopathia Striata with Cranial Sclerosis due to a de novo heterozygous nonsense mutation of the AMER1 gene.
Osteopathia Striata with Cranial Sclerosis (OS-CS), also known as Horan-Beighton Syndrome, is a rare genetic disease; about 90 cases have been reported to date. It is associated with mutations (heterozygous for female subjects and hemizygous for males) of the AMER1 gene, located at Xq11.2, and shows an X-linked pattern of transmission. Typical clinical manifestations include macrocephaly, characteristic facial features (frontal bossing, epicanthal folds, hypertelorism, depressed nasal bridge, orofacial cleft, prominent jaw), hearing loss and developmental delay. Males usually present a more severe phenotype than females and rarely survive. Diagnostic suspicion is based on clinical signs, radiographic findings of cranial and long bones sclerosis and metaphyseal striations, subsequent genetic testing may confirm it. Hereby, we report on a female newborn with frontal and parietal bossing, narrow bitemporal diameter, dysplastic, low-set and posteriorly rotated ears, microretrognathia, cleft palate, and rhizomelic shortening of lower limbs. Postnatally, she manifested feeding intolerance with biliary vomiting and abdominal distension. Therefore, in the suspicion of bowel obstruction, she underwent surgery, which evidenced and corrected an intestinal malrotation. Limbs X-ray and skull computed tomography investigations did not show cranial sclerosis and/or metaphyseal striations. Array-CGH analysis revealed normal findings. Then, a target next generation sequencing (NGS) analysis, including the genes involved in skeletal dysplasias, was performed and revealed a de novo heterozygous nonsense mutation of the AMER1 gene. The patient was discharged at 2 months of age and included in a multidisciplinary follow-up. Aged 9 months, she now shows developmental and growth (except for relative macrocephaly) delay. The surgical correction of cleft palate has been planned. Our report shows the uncommon association of intestinal malrotation in a female newborn with OS-CS. It highlights that neonatologists have to consider such a diagnosis, even in absence of cranial sclerosis and long bones striations, as these usually appear over time. Other syndromes with cranial malformations and skeletal dysplasia must be included in the differential diagnosis. The phenotypic spectrum is wide and variable in both genders. Due to variable X-inactivation, females may also show a severe and early-onset clinical picture. Multidisciplinary management and careful, early and long-term follow-up should be offered to these patients, in order to promptly identify any associated morbidities and prevent possible complications or adverse outcomes.
Wilms tumor in patients with osteopathia striata with cranial sclerosis.
Germline pathogenic variants in AMER1 cause osteopathia striata with cranial sclerosis (OSCS: OMIM 300373), an X-linked sclerosing bone disorder. Female heterozygotes exhibit metaphyseal striations in long bones, macrocephaly, cleft palate, and, occasionally, learning disability. Male hemizygotes typically manifest the condition as fetal or neonatal death. Somatically acquired variants in AMER1 are found in neoplastic tissue in 15-30% of patients with Wilms tumor; however, to date, only one individual with OSCS has been reported with a Wilms tumor. Here we present four cases of Wilms tumor in unrelated individuals with OSCS, including the single previously published case. We also report the first case of bilateral Wilms tumor in a patient with OSCS. Tumor tissue analysis showed no clear pattern of histological subtypes. In Beckwith-Wiedemann syndrome, which has a known predisposition to Wilms tumor development, clinical protocols have been developed for tumor surveillance. In the absence of further evidence, we propose a similar protocol for patients with OSCS to be instituted as an initial precautionary approach to tumor surveillance. Further evidence is needed to refine this protocol and to evaluate the possibility of development of other neoplasms later in life, in patients with OSCS.
First case of osteopathia striata with cranial sclerosis in an adult male with Klinefelter syndrome.
Osteopathia striata with cranial sclerosis is a rare X-linked disorder. It is often lethal in male patients, and is considered X-linked dominant since affected females exhibit clinical signs, although milder than males. We describe here an adult male patient, with clinical and radiological signs similar to those described in female patients. Diagnosis was confirmed by the identification of an AMER1 mutation. The presence of long bones striation and the clinical phenotype of the patient also led to the diagnosis of non-mosaic Klinefelter syndrome, probably explaining the non-lethal and even rather minor phenotype compared to the rare affected males already described.
Publicações recentes
Osteopathia striata-cranial sclerosis: otorhinolaryngologic clinical presentation and radiologic findings.
Comments on "osteopathia striata cranial sclerosis: non-random X-inactivation suggestive of X-linked dominant inheritance".
Conductive hearing loss in osteopathia striata-cranial sclerosis.
Osteopathia striata cranial sclerosis: non-random X-inactivation suggestive of X-linked dominant inheritance.
[Osteopathia striata-cranial sclerosis-megalencephaly].
📚 EuropePMCmostrando 4
Osteopathia striata with cranial sclerosis as a cancer predisposition syndrome: The first report of neuroblastoma and review of all cancers in OSCS.
American journal of medical genetics. Part AIntestinal malrotation in a female newborn affected by Osteopathia Striata with Cranial Sclerosis due to a de novo heterozygous nonsense mutation of the AMER1 gene.
Italian journal of pediatricsWilms tumor in patients with osteopathia striata with cranial sclerosis.
European journal of human genetics : EJHGFirst case of osteopathia striata with cranial sclerosis in an adult male with Klinefelter syndrome.
Joint bone spineAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de osteopatia estriada-esclerose craniana.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de osteopatia estriada-esclerose craniana
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Osteopathia striata with cranial sclerosis as a cancer predisposition syndrome: The first report of neuroblastoma and review of all cancers in OSCS.
- Intestinal malrotation in a female newborn affected by Osteopathia Striata with Cranial Sclerosis due to a de novo heterozygous nonsense mutation of the AMER1 gene.
- Wilms tumor in patients with osteopathia striata with cranial sclerosis.
- First case of osteopathia striata with cranial sclerosis in an adult male with Klinefelter syndrome.
- Osteopathia striata-cranial sclerosis: otorhinolaryngologic clinical presentation and radiologic findings.
- Comments on "osteopathia striata cranial sclerosis: non-random X-inactivation suggestive of X-linked dominant inheritance".
- Conductive hearing loss in osteopathia striata-cranial sclerosis.
- Osteopathia striata cranial sclerosis: non-random X-inactivation suggestive of X-linked dominant inheritance.
- [Osteopathia striata-cranial sclerosis-megalencephaly].
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2780(Orphanet)
- OMIM OMIM:300373(OMIM)
- MONDO:0010310(MONDO)
- GARD:4148(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q32136756(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome de osteopatia estriada-esclerose craniana
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata