Distúrbio raro de armazenamento lisossômico caracterizado bioquimicamente por atividade deficiente de beta-galactosidase e clinicamente por uma ampla gama de características neuroviscerais, oftalmológicas e dismórficas variáveis.
Introdução
O que você precisa saber de cara
Distúrbio raro de armazenamento lisossômico caracterizado bioquimicamente por atividade deficiente de beta-galactosidase e clinicamente por uma ampla gama de características neuroviscerais, oftalmológicas e dismórficas variáveis.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 47 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 154 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisCleaves beta-linked terminal galactosyl residues from gangliosides, glycoproteins, and glycosaminoglycans Has no beta-galactosidase catalytic activity, but plays functional roles in the formation of extracellular elastic fibers (elastogenesis) and in the development of connective tissue. Seems to be identical to the elastin-binding protein (EBP), a major component of the non-integrin cell surface receptor expressed on fibroblasts, smooth muscle cells, chondroblasts, leukocytes, and certain cance
LysosomeCytoplasm, perinuclear region
GM1-gangliosidosis 1
An autosomal recessive lysosomal storage disease marked by the accumulation of GM1 gangliosides, glycoproteins and keratan sulfate primarily in neurons of the central nervous system. GM1-gangliosidosis type 1 is characterized by onset within the first three months of life, central nervous system degeneration, coarse facial features, hepatosplenomegaly, skeletal dysmorphology reminiscent of Hurler syndrome, and rapidly progressive psychomotor deterioration. Urinary oligosaccharide levels are high. It leads to death usually between the first and second year of life.
Variantes genéticas (ClinVar)
389 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 1.059 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
8 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Gangliosidose GM1
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
6 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
30 ensaios clínicos encontrados, 8 ativos.
Publicações mais relevantes
AAV9 Gene Therapy in Type II GM1 Gangliosidosis - A Phase 1-2 Trial.
GM1 gangliosidosis, caused by biallelic variants in GLB1, results from deficiency of lysosomal β-galactosidase, which degrades GM1 ganglioside. This fatal neurodegenerative disease currently has no effective therapy. In a phase 1-2, open-label, dose-escalation study, we assessed immunosuppression and a single intravenous infusion of adeno-associated virus serotype 9 (AAV9) encoding β-galactosidase in children with type II GM1 gangliosidosis with late-infantile or juvenile onset. The primary end point was safety. Secondary end points included changes from baseline in the cerebrospinal fluid (CSF) GM1 ganglioside concentration and β-galactosidase activity, clinical assessments (including the Clinical Global Impression-Improvement [CGI-I] score, assessed on a scale from 1 [very much improved] to 7 [very much worse]), and neuroimaging patterns. Nine participants were enrolled. Over a 3-year period, 124 adverse events occurred, 30 of which (8 gastrointestinal events, 21 laboratory abnormalities associated with inflammation, and 1 tachycardia event) were deemed by the investigator as being possibly, probably, or definitely related to the gene therapy. Five serious adverse events occurred, including vomiting that led to hospitalization in one participant, which was attributed to the gene therapy. Serum aspartate and alanine aminotransferase levels increased in all participants and returned to baseline levels by 18 months. In all participants, the CSF β-galactosidase level increased and CSF GM1 ganglioside level decreased. Expressive communication and gross motor scores appeared stable, but fine motor and receptive communication scores decreased. The median CGI-I score was 3 (indicating minimal improvement) at 2 years and 4 (indicating no change) at 3 years; in historical controls, scores have been shown to increase (indicating worsening) over time. Neuroimaging showed patterns consistent with reduced rates of cerebral atrophy and favorable changes in myelination as compared with baseline. In this study involving nine participants with type II GM1 gangliosidosis, a single infusion of AAV9 encoding β-galactosidase was associated with adverse events, including severe vomiting in one participant and elevated liver-enzyme levels in all participants. Secondary end-point results suggested improvements in biochemical markers and neuroimaging patterns and stable or reduced rates of developmental deterioration in some measures. (Funded by the National Human Genome Research Institute and others; ClinicalTrials.gov number, NCT03952637.).
From Molecule to Meaning: Neuronopathic Biomarkers and Clinical Relevance in GM1.
GM1 gangliosidosis is a rare, progressively neurodegenerative lysosomal storage disorder characterized by profound central nervous system involvement and substantial clinical heterogeneity. The development of reliable biomarkers is essential for tracking disease progression, stratifying patients, and advancing clinical trial readiness. Primary substrate markers, including cerebrospinal fluid (CSF) GM1 ganglioside and related lysosphingolipids, provide direct biochemical indices of lysosomal dysfunction. Emerging glycan signatures may further reflect disruptions in glycosylation pathways and responses to therapeutic intervention. Neurofilament light chain (NfL), a sensitive indicator of axonal injury, is consistently elevated in GM1 and shows promise as a fluid biomarker, although it does not convey regional specificity. Neuroimaging offers complementary insight into the structural and metabolic consequences of disease. Characteristic findings include diffuse white matter abnormalities, thalamic and basal ganglia signal changes, cortical and cerebellar atrophy, and ventriculomegaly. Quantitative MRI and magnetic resonance spectroscopy (MRS) reveal longitudinal declines in tissue volume and neuronal integrity that parallel functional deterioration. Diffusion-based methods, including differential tractography, highlight progressive loss of white matter microstructure relative to age-matched controls. However, severe anatomical distortion in GM1 limits the applicability of standard neuroimaging atlases, necessitating manual or semi-automated segmentation approaches. Together, fluid biomarkers (gangliosides, lysosphingolipids, glycans, NfL) and advanced neuroimaging metrics (volumetry, MRS, diffusion imaging) establish a multimodal framework for evaluating disease burden and therapeutic response. Standardized methodologies, harmonized natural history datasets, and genotype-stratified analyses will be critical for validating these biomarkers across GM1 subtypes. Strengthening this biomarker ecosystem will enable sensitive and clinically meaningful endpoints to support future therapeutic development in GM1 gangliosidosis.
Lentiviral hematopoietic stem cell gene therapy ameliorates GM1-gangliosidosis in mice.
Lysosomal storage diseases (LSDs) are >70 devasting diseases causing progressive neurodegeneration and systemic symptoms. Hematopoietic stem cell (HSC) gene therapy (GT), i.e., the autologous transplantation of genetically corrected HSCs by lentiviral transduction, has shown transformative efficacy in multiple neurodegenerative LSDs so far. We developed a translational HSC GT approach for GM1-gangliosidosis, a neurodegenerative LSD due to β-galactosidase (β-gal) deficiency, based on a lentiviral vector expressing either the human or the murine therapeutic enzyme. Cells were administered by a standard intravenous injection, or by an innovative, combined systemic and local cell delivery to enhance the GT efficacy. In the murine model of the disease, GT broadly restored enzymatic activity and reduced the lysosomal storage in the brain, ameliorated the neuromuscular phenotype, and increased animal survival. Combined cell-delivery GT with the human enzyme, or standard intravenous GT with the murine β-gal were similarly effective in mitigating disease phenotype, indicating a substantial therapeutic potential of the approach for a future clinical translation.
Alkaline Phosphatase and Infantile GM1 Gangliosidosis: A Simple Biomarker for a Complex Disease?
GM1 gangliosidosis is a lysosomal storage disease (LSD) caused by β-galactosidase deficiency, characterized by the accumulation of gangliosides in various tissues. Among different GM1 forms (infantile form, late-infantile and juvenile form, and late-onset form), the infantile form is the most severe: despite an early clinical onset with rapid neurodegeneration, coarse face, abdominal visceromegaly and skeletal abnormalities, the diagnosis is usually delayed, given the lack of recognized early disease-specific markers. We report the case of a newborn presenting with mild edema of hands and feet, mild transient hypoalbuminemia and isolated hyperphosphatasemia at three weeks of life. The first cardiological evaluation showed mild mitral regurgitation. Despite the absence of neurological symptoms, organomegaly, or a coarse face, the turgid consistency of the limbs, together with mitral regurgitation and persistent hyperphosphatasemia, led to multiorgan investigations with discovery of bilateral cherry-red spots and a beak-shaped lumbar vertebra. The cardiological follow-up revealed a dysplastic mitral valve. In the suspicion of a lysosomal disease, biochemical investigations were planned. An altered profile of urinary oligosaccharides, along with low β-galactosidase activity in leukocytes, led to the diagnosis of infantile GM1 gangliosidosis at 3 months of age. The GLB1 gene analysis confirmed the diagnosis. Genetic testing for GLB1 should be considered in cases of persistent hyperphosphatemia, especially if it is associated with any other clinical indicator of GM1, such as limb edema.
Novel Galactosidase-Beta-1 Variant in Infantile GM1 Gangliosidosis: A Case Report.
GM1 gangliosidosis is an autosomal recessive lysosomal storage disorder caused by pathogenic GLB1 variants that impair β-galactosidase activity, resulting in GM1 ganglioside accumulation. The infantile (type I) form is the most severe. We describe the case of a one-year-old girl born to consanguineous parents who presented with developmental regression, hypotonia, coarse facial features, hepatosplenomegaly, macular cherry-red spots, Mongolian spots, and sensorineural hearing loss. Whole-exome sequencing revealed a novel homozygous GLB1 variant, NM_000404.4:c.1525T>A (p.Trp509Arg), absent from population databases and predicted deleterious by in silico tools. According to the American College of Medical Genetics and Genomics guidelines, it is classified as a variant of uncertain significance, though the phenotype-genotype match suggests pathogenicity. This case broadens the GLB1 mutational spectrum and underscores the value of early genetic testing for diagnosis, counseling, and management.
Publicações recentes
Alkaline Phosphatase and Infantile GM1 Gangliosidosis: A Simple Biomarker for a Complex Disease?
Novel Galactosidase-Beta-1 Variant in Infantile GM1 Gangliosidosis: A Case Report.
Clinical, Radiological, and Genetic Profiles of Eight Patients with Combined Dystonic Manifestation of Type-III GM1 Gangliosidosis: A Video Case Series from India.
AAV9 Gene Therapy in Type II GM1 Gangliosidosis - A Phase 1-2 Trial.
Bioresponsive MR Imaging Probes for Noninvasive Monitoring of AAV Gene Therapy.
📚 EuropePMC459 artigos no totalmostrando 197
Alkaline Phosphatase and Infantile GM1 Gangliosidosis: A Simple Biomarker for a Complex Disease?
JIMD reportsNovel Galactosidase-Beta-1 Variant in Infantile GM1 Gangliosidosis: A Case Report.
CureusClinical, Radiological, and Genetic Profiles of Eight Patients with Combined Dystonic Manifestation of Type-III GM1 Gangliosidosis: A Video Case Series from India.
Tremor and other hyperkinetic movements (New York, N.Y.)AAV9 Gene Therapy in Type II GM1 Gangliosidosis - A Phase 1-2 Trial.
The New England journal of medicineBioresponsive MR Imaging Probes for Noninvasive Monitoring of AAV Gene Therapy.
Bioconjugate chemistryFrom Molecule to Meaning: Neuronopathic Biomarkers and Clinical Relevance in GM1.
Journal of inherited metabolic diseaseLentiviral hematopoietic stem cell gene therapy ameliorates GM1-gangliosidosis in mice.
Molecular therapy : the journal of the American Society of Gene TherapyA novel HEXA frameshift mutation identified in Angus cattle with GM2 gangliosidosis.
Animal geneticsBurden of caregiving of individuals with GM1 and GM2 gangliosidoses in the United States: a qualitative study.
Orphanet journal of rare diseasesCongenital Dermal Melanocytosis Exhibited in Two Patients with Hurler Syndrome: Clinical Characterization and Report of a Recurrent IDUA Allele in Colombia.
International journal of molecular sciencesCritical Functional Domains in Pediatric Onset TUBB4A-Related Leukodystrophy: A Clinical and Caregiver's Perspective.
Pediatric neurologyLysosomal Network Defects in Early-Onset Parkinson's Disease Patients Carrying Rare Variants in Lysosomal Hydrolytic Enzyme Genes.
International journal of molecular sciencesNeuroimaging Spectrum of GM1 Gangliosidosis with Description of Novel Imaging Signs.
AJNR. American journal of neuroradiologyNovel insights into pathomechanisms of retinal neuronal degeneration and reactive gliosis in a murine model of GM1-gangliosidosis.
Scientific reportsAAV9 Gene Therapy in GM1 Gangliosidosis Type II: A Phase 1/2 Trial.
medRxiv : the preprint server for health sciencesβ-Galactosidase inhibition explored by biochemical methods and in silico studies for plant polyphenols.
Archives of biochemistry and biophysicsPlasma membrane remodeling in GM2 gangliosidoses drives synaptic dysfunction.
PLoS biologySecondary accumulation of lyso-platelet activating factors in lysosomal storage diseases.
Molecular genetics and metabolismDifferential tractography: an imaging marker for tissue degeneration in neurodegenerative diseases.
Brain communicationsCorrigendum: Infection of a β-galactosidase-deficient mouse strain with Theiler's murine encephalomyelitis virus reveals limited immunological dysregulations in this lysosomal storage disease.
Frontiers in immunologyBrain Age Prediction in Type II GM1 Gangliosidosis.
medRxiv : the preprint server for health sciencesInfection of a β-galactosidase-deficient mouse strain with Theiler's murine encephalomyelitis virus reveals limited immunological dysregulations in this lysosomal storage disease.
Frontiers in immunologyA Case for Automated Segmentation of MRI Data in Neurodegenerative Diseases: Type II GM1 Gangliosidosis.
NeuroSciCorrigendum to "Natural history progression of MRI brain volumetrics in type II late-infantile and juvenile GM1 gangliosidosis patients" [Molecular Genetics and Metabolism 2025 Mar;144(3):109025].
Molecular genetics and metabolismNovel Pathogenic Variants in POLR3K Cause POLR3-Related Leukodystrophy.
Human mutationAdeno-associated virus expressing a blood-brain barrier-penetrating enzyme improves GM1 gangliosidosis in a preclinical model.
The Journal of clinical investigationImaging manifestations in infantile GM1 gangliosidosis: a rare lysosomal storage disorder: a paediatric case report.
BJR case reportsRetrospective assessment of clinical global impression of severity and change in GM1 gangliosidosis: a tool to score natural history data in rare disease cohorts.
Orphanet journal of rare diseasesA Case for Automated Segmentation of MRI Data in Milder Neurodegenerative Diseases.
medRxiv : the preprint server for health sciencesEvaluation of the Landscape of Pharmacodynamic Biomarkers in GM1 and GM2 Gangliosidosis.
Clinical and translational scienceClinical and genetic analysis of a Chinese family with GM1 gangliosidosis caused by a novel mutation in GLB1 gene.
Frontiers in pediatricsPersistent elevations of alkaline phosphatase as an early indicator of GM1 gangliosidosis.
Molecular genetics and metabolism reportsNatural history progression of MRI brain volumetrics in type II late-infantile and juvenile GM1 gangliosidosis patients.
Molecular genetics and metabolismA natural history study of pediatric patients with early onset of GM1 gangliosidosis, GM2 gangliosidoses, or gaucher disease type 2 (RETRIEVE).
Orphanet journal of rare diseasesDifferential Tractography: A Biomarker for Neuronal Function in Neurodegenerative Disease.
medRxiv : the preprint server for health sciencesQuantitative reliability assessment of brain MRI volumetric measurements in type II GM1 gangliosidosis patients.
Frontiers in neuroimagingGeneration of an infantile GM1 gangliosidosis induced pluripotent stem cell line (CHOCi005-A) for disease modeling and therapeutic testing.
Stem cell researchBase editing of the GLB1 gene is therapeutic in GM1 gangliosidosis patient-derived cells.
Molecular genetics and metabolismValidation of high-sensitivity assays to quantitate cerebrospinal fluid and serum β-galactosidase activity in patients with GM1-gangliosidosis.
Molecular therapy. Methods & clinical developmentInsights into the Pathobiology of GM1 Gangliosidosis from Single-Nucleus Transcriptomic Analysis of CNS Cells in a Mouse Model.
International journal of molecular sciencesHybrid lipid-AuNP clusters as highly efficient SERS substrates for biomedical applications.
Nature communicationsEstablishment of iPS cell line (SDQLCHi080-A) from a patient with GM1 gangliosidosis due to GLB1 mutation.
Stem cell researchCreation of an in vitro model of GM1 gangliosidosis by CRISPR/Cas9 knocking-out the GLB1 gene in SH-SY5Y human neuronal cell line.
Cell biochemistry and functionTherapeutic developments for neurodegenerative GM1 gangliosidosis.
Frontiers in neuroscienceInherited metabolic disorders in Cyprus.
Molecular genetics and metabolism reportsGM1 gangliosidosis type II: Results of a 10-year prospective study.
Genetics in medicine : official journal of the American College of Medical GeneticsAltered GM1 catabolism affects NMDAR-mediated Ca2+ signaling at ER-PM junctions and increases synaptic spine formation in a GM1-gangliosidosis model.
Cell reportsGM1 and GM2-Gangliosidosis: Clinical Features, Neuroimaging Findings and Electroencephalography.
Iranian journal of child neurologyMorquio B disease: a case report.
Frontiers in pediatricsCase report: Preimplantation genetic testing for infantile GM1 gangliosidosis.
Frontiers in geneticsIdentification of GM1-Ganglioside Secondary Accumulation in Fibroblasts from Neuropathic Gaucher Patients and Effect of a Trivalent Trihydroxypiperidine Iminosugar Compound on Its Storage Reduction.
Molecules (Basel, Switzerland)Extensive and Persistent Dermal Melanocytosis in a Male Carrier of Mucopolysaccharidosis Type IIIC (Sanfilippo Syndrome): A Case Report.
Children (Basel, Switzerland)Extensive Telangiectasias and Mongolian Spots: A Clue towards GM1-Gangliosidosis.
Indian journal of dermatologyA Strategic Translational Research System for Drug Discovery in Tottori University.
Yonago acta medicaGene therapy approaches for GM1 gangliosidosis: Focus on animal and cellular studies.
Cell biochemistry and functionBiallelic pathogenic variants in POLR3D alter tRNA transcription and cause a hypomyelinating leukodystrophy: A case report.
Frontiers in neurologySialidase NEU3 action on GM1 ganglioside is neuroprotective in GM1 gangliosidosis.
Journal of lipid researchThe development of a broad-spectrum retaining β-exo-galactosidase activity-based probe.
Organic & biomolecular chemistryMongolian spots in a sick patient-A diagnosis of infantile GM1 gangliosidosis.
The Australasian journal of dermatologyFrom amaurotic idiocy to biochemically defined lipid storage diseases: the first identification of GM1-Gangliosidosis.
Free neuropathologyAltered GM1 catabolism affects NMDAR-mediated Ca 2+ signaling at ER-PM junctions and increases synaptic spine formation.
bioRxiv : the preprint server for biologyHypomyelination caused by a novel homozygous pathogenic variant in FOLR1: complete clinical and radiological recovery with oral folinic acid therapy and review of the literature.
Orphanet journal of rare diseasesEndogenous, non-reducing end glycosaminoglycan biomarkers are superior to internal disaccharide glycosaminoglycan biomarkers for newborn screening of mucopolysaccharidoses and GM1 gangliosidosis.
Molecular genetics and metabolismNatural history of GM1 gangliosidosis-Retrospective cohort study of 61 French patients from 1998 to 2019.
Journal of inherited metabolic diseaseGanglioside GM1 and the Central Nervous System.
International journal of molecular sciencesA pentasaccharide for monitoring pharmacodynamic response to gene therapy in GM1 gangliosidosis.
EBioMedicineFetal Homozygous GM1 Gangliosidosis Presenting as Transient Non-immune Hydrops Fetalis.
The Israel Medical Association journal : IMAJThe Experience of Parents of Children With Genetically Determined Leukoencephalopathies With the Health Care System: A Qualitative Study.
Journal of child neurologyCraniofacial features of POLR3-related leukodystrophy caused by biallelic variants in POLR3A, POLR3B and POLR1C.
Journal of medical geneticsSolving inherited white matter disorder etiologies in the neurology clinic: Challenges and lessons learned using next-generation sequencing.
Frontiers in neurologyGM1 gangliosidosis: patients with different phenotypic features and novel mutations.
Journal of pediatric endocrinology & metabolism : JPEMGene Therapy of Sphingolipid Metabolic Disorders.
International journal of molecular sciencesAnalysis of urinary oligosaccharide excretion patterns by UHPLC/HRAM mass spectrometry for screening of lysosomal storage disorders.
Journal of inherited metabolic diseaseGlb1 knockout mouse model shares natural history with type II GM1 gangliosidosis patients.
Molecular genetics and metabolismThe Antifungal Antibiotic Filipin as a Diagnostic Tool of Cholesterol Alterations in Lysosomal Storage Diseases and Neurodegenerative Disorders.
Antibiotics (Basel, Switzerland)Gene Editing Corrects In Vitro a G > A GLB1 Transition from a GM1 Gangliosidosis Patient.
The CRISPR journalGM1-gangliosidosis: The caregivers' assessments of symptom impact and most important symptoms to treat.
American journal of medical genetics. Part AAnesthesia outcomes in lysosomal disorders: CLN3 and GM1 gangliosidosis.
American journal of medical genetics. Part Aβ-Galactosidase deficiency in the GLB1 spectrum of lysosomal storage disease can present with severe muscle weakness and atrophy.
JIMD reportsAAVrh10 vector corrects pathology in animal models of GM1 gangliosidosis and achieves widespread distribution in the CNS of nonhuman primates.
Molecular therapy. Methods & clinical developmentPreclinical Enzyme Replacement Therapy with a Recombinant β-Galactosidase-Lectin Fusion for CNS Delivery and Treatment of GM1-Gangliosidosis.
CellsFluorescent In Situ Staining and Flow Cytometric Procedures as New Pre-Diagnostic Tests for Sialidosis, GM1 Gangliosidosis and Niemann-Pick Type C.
BiomedicinesSingle Institutional Experience with GM1 Gangliosidosis: Clinical and Laboratory Results of 14 Patients.
Balkan medical journalAbnormal DaTscan in GM1-Gangliosidosis Type III Manifesting with Dystonia-Parkinsonism.
Movement disorders clinical practiceSynthesis of a New β-Galactosidase Inhibitor Displaying Pharmacological Chaperone Properties for GM1 Gangliosidosis.
Molecules (Basel, Switzerland)Carrier Rate and Mutant Allele Frequency of GM1 Gangliosidosis in Miniature Shiba Inus (Mame Shiba): Population Screening of Breeding Dogs in Japan.
Animals : an open access journal from MDPIHematopoietic stem cell gene therapy ameliorates CNS involvement in murine model of GM1-gangliosidosis.
Molecular therapy. Methods & clinical development[Genetic and clinical analysis of a novel GLB1 gene variant in a Chinese patient with GM1-gangliosidosis].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsRare Diseases in Glycosphingolipid Metabolism.
Advances in experimental medicine and biologyAAV9-coGLB1 Improves Lysosomal Storage and Rescues Central Nervous System Inflammation in a Mutant Mouse Model of GM1 Gangliosidosis.
Current gene therapyPlasma neurofilament light, glial fibrillary acidic protein and lysosphingolipid biomarkers for pharmacodynamics and disease monitoring of GM2 and GM1 gangliosidoses patients.
Molecular genetics and metabolism reportsLate-infantile GM1 gangliosidosis: A case report.
MedicineLysosomal Storage Disorders: Clinical, Biochemical and molecular profile from Rare disease centre, India.
Annals of Indian Academy of NeurologyPositioning Head Tilt in Canine Lysosomal Storage Disease: A Retrospective Observational Descriptive Study.
Frontiers in veterinary science[Increased use of genetic health care in Iceland 2012-2017].
LaeknabladidPhenotypical changes of satellite glial cells in a murine model of GM1 -gangliosidosis.
Journal of cellular and molecular medicineReal-time MR tracking of AAV gene therapy with βgal-responsive MR probe in a murine model of GM1-gangliosidosis.
Molecular therapy. Methods & clinical developmentChitotriosidase as a biomarker for gangliosidoses.
Molecular genetics and metabolism reportsGM1 Gangliosidosis-A Mini-Review.
Frontiers in geneticsCommentary: GM1-Gangliosidosis Type III Associated Parkinsonism.
Movement disorders clinical practiceGM1-Gangliosidosis Type III Associated Parkinsonism.
Movement disorders clinical practiceWhite Matter Pathology as a Barrier to Gangliosidosis Gene Therapy.
Frontiers in cellular neuroscienceIntravenous delivery of adeno-associated viral gene therapy in feline GM1 gangliosidosis.
Brain : a journal of neurologyEstimating the prevalence of Niemann-Pick disease type C (NPC) in the United States.
Molecular genetics and metabolismA 7-month-old boy with failure to thrive.
Brain pathology (Zurich, Switzerland)Morquio-like dysostosis multiplex presenting with neuronopathic features is a distinct GLB1-related phenotype.
JIMD reportsLysosomal storage disorders as an etiology of nonimmune hydrops fetalis: A systematic review.
Clinical geneticsStructure of the murine lysosomal multienzyme complex core.
Science advancesGM1 Gangliosidosis: Mechanisms and Management.
The application of clinical geneticsExamination of a blood-brain barrier targeting β-galactosidase-monoclonal antibody fusion protein in a murine model of GM1-gangliosidosis.
Molecular genetics and metabolism reportsRapid Identification of New Biomarkers for the Classification of GM1 Type 2 Gangliosidosis Using an Unbiased 1H NMR-Linked Metabolomics Strategy.
CellsDisentangling molecular and clinical stratification patterns in beta-galactosidase deficiency.
Journal of medical geneticsA GM1 gangliosidosis mutant mouse model exhibits activated microglia and disturbed autophagy.
Experimental biology and medicine (Maywood, N.J.)Morquio B disease: From pathophysiology towards diagnosis.
Molecular genetics and metabolismA computational approach to analyse the amino acid variants of GLB1 protein causing GM1 Gangliosidosis.
Metabolic brain diseaseMorquio B Disease. Disease Characteristics and Treatment Options of a Distinct GLB1-Related Dysostosis Multiplex.
International journal of molecular sciencesThe juvenile gangliosidoses: A timeline of clinical change.
Molecular genetics and metabolism reportsClinical and Laboratory Profile of Gangliosidosis from Southern Part of India.
Journal of pediatric geneticsA Single Injection of an Optimized Adeno-Associated Viral Vector into Cerebrospinal Fluid Corrects Neurological Disease in a Murine Model of GM1 Gangliosidosis.
Human gene therapyMongolian spots in GM1 gangliosidosis: a pictorial report.
Clinical dysmorphologyMechanistic Insights into the Chaperoning of Human Lysosomal-Galactosidase Activity: Highly Functionalized Aminocyclopentanes and C-5a-Substituted Derivatives of 4-epi-Isofagomine.
Molecules (Basel, Switzerland)Substrate reduction therapy with Miglustat in pediatric patients with GM1 type 2 gangliosidosis delays neurological involvement: A multicenter experience.
Molecular genetics & genomic medicineIntermittent enzyme replacement therapy with recombinant human β-galactosidase prevents neuraminidase 1 deficiency.
The Journal of biological chemistryThe Lysosomal Diseases Testing Laboratory: A review of the past 47 years.
JIMD reportsDetection of GM1-gangliosidosis in newborn dried blood spots by enzyme activity and biomarker assays using tandem mass spectrometry.
Journal of inherited metabolic diseaseUtility of metabolic screening in neurological presentations of infancy.
Annals of clinical and translational neurologyNovel Drug Candidates Improve Ganglioside Accumulation and Neural Dysfunction in GM1 Gangliosidosis Models with Autophagy Activation.
Stem cell reportsAxonopathy and Reduction of Membrane Resistance: Key Features in a New Murine Model of Human GM1-Gangliosidosis.
Journal of clinical medicineAn autopsy case of GM1 gangliosidosis type II in a patient who survived a long duration with artificial respiratory support.
Neuropathology : official journal of the Japanese Society of NeuropathologyThe pharmacological chaperone N-n-butyl-deoxygalactonojirimycin enhances β-galactosidase processing and activity in fibroblasts of a patient with infantile GM1-gangliosidosis.
Human geneticsLysosomal storage disease spectrum in nonimmune hydrops fetalis: a retrospective case control study.
Prenatal diagnosisEarly detection of lysosomal diseases by screening of cases of idiopathic splenomegaly and/or thrombocytopenia with a next-generation sequencing gene panel.
JIMD reportsGenetic Base of Behavioral Disorders in Mucopolysaccharidoses: Transcriptomic Studies.
International journal of molecular sciences7T MRI Predicts Amelioration of Neurodegeneration in the Brain after AAV Gene Therapy.
Molecular therapy. Methods & clinical developmentThe natural history of Type 1 infantile GM1 gangliosidosis: A literature-based meta-analysis.
Molecular genetics and metabolismBi-functional IgG-lysosomal enzyme fusion proteins for brain drug delivery.
Scientific reportsPre-diagnosing and managing patients with GM1 gangliosidosis and related disorders by the evaluation of GM1 ganglioside content.
Scientific reportsAdvances in Sphingolipidoses: CRISPR-Cas9 Editing as an Option for Modelling and Therapy.
International journal of molecular sciencesThe Clinical and Molecular Spectrum of GM1 Gangliosidosis.
The Journal of pediatricsCharacterization of glycan substrates accumulating in GM1 Gangliosidosis.
Molecular genetics and metabolism reportsThe skeletal phenotype of intermediate GM1 gangliosidosis: Clinical, radiographic and densitometric features, and implications for clinical monitoring and intervention.
BoneProgressive dystonia as a presenting manifestation of GM1 gangliosidosis.
Clinical parkinsonism & related disordersHuman GLB1 knockout cerebral organoids: A model system for testing AAV9-mediated GLB1 gene therapy for reducing GM1 ganglioside storage in GM1 gangliosidosis.
Molecular genetics and metabolism reportsClinical findings in Brazilian patients with adult GM1 gangliosidosis.
JIMD reportsIntracerebroventricular enzyme replacement therapy with β-galactosidase reverses brain pathologies due to GM1 gangliosidosis in mice.
The Journal of biological chemistryGenotype-phenotype correlation of gangliosidosis mutations using in silico tools and homology modeling.
Molecular genetics and metabolism reportsTeaching NeuroImages: Wishbone pattern of iron accumulation: A characteristic imaging sign in GM1 gangliosidosis.
NeurologyA possible biomarker of neurocytolysis in infantile gangliosidoses: aspartate transaminase.
Metabolic brain disease[Novel mutations of GLB1 gene identified in a Chinese pedigree affected with GM1 gangliosidosis].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics[Identification and pathogenicity prediction of a novel GLB1 variant c.101T>C (p.Ile34Thr) in an infant with GM1 gangliosidosis].
Zhongguo dang dai er ke za zhi = Chinese journal of contemporary pediatricsPathological findings of central nervous system, two GM1 gangliosidosis autopsy cases.
The Turkish journal of pediatricsGenetic Analysis of Undiagnosed Juvenile GM1-Gangliosidosis by Microarray and Exome Sequencing.
Case reports in geneticsIdentification of a novel GLB1 mutation in a consanguineous Pakistani family affected by rare infantile GM1 gangliosidosis.
Journal of geneticsComprehensive behavioral and biochemical outcomes of novel murine models of GM1-gangliosidosis and Morquio syndrome type B.
Molecular genetics and metabolismVacuolated Lymphocytes as a Clue for Diagnosis of Lysosomal Storage Disease like GM1 Gangliosidosis.
Indian journal of hematology & blood transfusion : an official journal of Indian Society of Hematology and Blood TransfusionNeonatal punctate calcifications associated with maternal mixed connective tissue disorder (MCTD).
BMJ case reportsExtensive Mongolian Spots in a Hypotonic Infant with GM1 Gangliosidosis.
Journal of pediatric neurosciences4-epi-Isofagomine derivatives as pharmacological chaperones for the treatment of lysosomal diseases linked to β-galactosidase mutations: Improved synthesis and biological investigations.
Bioorganic & medicinal chemistryClinical and molecular characteristics of 11 Chinese probands with GM1 gangliosidosis.
Metabolic brain diseaseProtein modeling and clinical description of a novel in-frame GLB1 deletion causing GM1 gangliosidosis type II.
Molecular genetics & genomic medicineMorquio B patient/caregiver survey: First insight into the natural course of a rare GLB1 related condition.
Molecular genetics and metabolism reportsIntracellular Delivery of β-Galactosidase Enzyme Using Arginase-Responsive Dextran Sulfate/Poly-l-arginine Capsule for Lysosomal Storage Disorder.
ACS omegaTeaching NeuroImages: Brain MRI and DaT-SPECT imaging in adult GM1 gangliosidosis.
NeurologyCysts in White Matter: A Novel Neuroimaging Finding in Infantile GM1 Gangliosidosis.
Annals of Indian Academy of NeurologyExpanding the phenotypic spectrum of type III GM1 gangliosidosis: Progressive dystonia with auditory startle.
Neurology IndiaMitochondria-associated membranes in aging and senescence: structure, function, and dynamics.
Cell death & diseaseMetabolic causes of nonimmune hydrops fetalis: A next-generation sequencing panel as a first-line investigation.
Clinica chimica acta; international journal of clinical chemistryHypomyelinating disorders in China: The clinical and genetic heterogeneity in 119 patients.
PloS oneDiagnostic challenge for the rare lysosomal storage disease: Late infantile GM1 gangliosidosis.
Brain & developmentSAAMP 2.0: An algorithm to predict genotype-phenotype correlation of lysosomal storage diseases.
Clinical geneticsDistinct progression patterns of brain disease in infantile and juvenile gangliosidoses: Volumetric quantitative MRI study.
Molecular genetics and metabolismMosaic uniparental disomy results in GM1 gangliosidosis with normal enzyme assay.
American journal of medical genetics. Part ALysosomal storage diseases.
Translational science of rare diseasesPolyethylene glycol-b-poly(lactic acid) polymersomes as vehicles for enzyme replacement therapy.
Nanomedicine (London, England)Canine GM2-Gangliosidosis Sandhoff Disease Associated with a 3-Base Pair Deletion in the HEXB Gene.
Journal of veterinary internal medicineRapid Targeted Genomics in Critically Ill Newborns.
PediatricsLipidomic Evaluation of Feline Neurologic Disease after AAV Gene Therapy.
Molecular therapy. Methods & clinical developmentLC-MS/MS multiplex analysis of lysosphingolipids in plasma and amniotic fluid: A novel tool for the screening of sphingolipidoses and Niemann-Pick type C disease.
PloS oneCase reports of juvenile GM1 gangliosidosisis type II caused by mutation in GLB1 gene.
BMC medical geneticsThe treatment of juvenile/adult GM1-gangliosidosis with Miglustat may reverse disease progression.
Metabolic brain diseaseLinking mitochondrial dysfunction to neurodegeneration in lysosomal storage diseases.
Journal of inherited metabolic diseaseInfantile gangliosidoses: Mapping a timeline of clinical changes.
Molecular genetics and metabolismDevelopment of a new tandem mass spectrometry method for urine and amniotic fluid screening of oligosaccharidoses.
Rapid communications in mass spectrometry : RCMNovel Biomarkers of Human GM1 Gangliosidosis Reflect the Clinical Efficacy of Gene Therapy in a Feline Model.
Molecular therapy : the journal of the American Society of Gene TherapyPeripheral blood findings in GM1 gangliosidosis.
BloodMongolian Spots in GM1 Gangliosidosis.
Indian pediatricsMultigene panel next generation sequencing in a patient with cherry red macular spot: Identification of two novel mutations in NEU1 gene causing sialidosis type I associated with mild to unspecific biochemical and enzymatic findings.
Molecular genetics and metabolism reports(5aR)-5a-C-Pentyl-4-epi-isofagomine: A powerful inhibitor of lysosomal β-galactosidase and a remarkable chaperone for mutations associated with GM1-gangliosidosis and Morquio disease type B.
European journal of medicinal chemistryDefining the genetic basis of early onset hereditary spastic paraplegia using whole genome sequencing.
NeurogeneticsThe GM1 and GM2 Gangliosidoses: Natural History and Progress toward Therapy.
Pediatric endocrinology reviews : PER4th Rare Disease South Eastern Europe (See) Meeting Skopje, Macedonia (November 14th, 2015).
Prilozi (Makedonska akademija na naukite i umetnostite. Oddelenie za medicinski nauki)Synthesis of C-5a-substituted derivatives of 4-epi-isofagomine: notable β-galactosidase inhibitors and activity promotors of GM1-gangliosidosis related human lysosomal β-galactosidase mutant R201C.
Carbohydrate researchPanthotenate Kinase-Associated Neurodegeneration Has a Founder Mutation (c.215_216insa) in Indian Agrawal Patients.
Movement disorders clinical practiceGlycomimetic-based pharmacological chaperones for lysosomal storage disorders: lessons from Gaucher, GM1-gangliosidosis and Fabry diseases.
Chemical communications (Cambridge, England)High-throughput imaging method for direct assessment of GM1 ganglioside levels in mammalian cells.
Data in briefSynthesis of C-5a-chain extended derivatives of 4-epi-isofagomine: Powerful β-galactosidase inhibitors and low concentration activators of GM1-gangliosidosis-related human lysosomal β-galactosidase.
Bioorganic & medicinal chemistry lettersVacuolated lymphocytes signifying a metabolic disorder in an infant with developmental delay.
Clinical case reportsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Gangliosidose GM1.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Gangliosidose GM1
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- AAV9 Gene Therapy in Type II GM1 Gangliosidosis - A Phase 1-2 Trial.
- From Molecule to Meaning: Neuronopathic Biomarkers and Clinical Relevance in GM1.
- Lentiviral hematopoietic stem cell gene therapy ameliorates GM1-gangliosidosis in mice.Molecular therapy : the journal of the American Society of Gene Therapy· 2026· PMID 41376163mais citado
- Alkaline Phosphatase and Infantile GM1 Gangliosidosis: A Simple Biomarker for a Complex Disease?
- Novel Galactosidase-Beta-1 Variant in Infantile GM1 Gangliosidosis: A Case Report.
- Clinical, Radiological, and Genetic Profiles of Eight Patients with Combined Dystonic Manifestation of Type-III GM1 Gangliosidosis: A Video Case Series from India.
- Bioresponsive MR Imaging Probes for Noninvasive Monitoring of AAV Gene Therapy.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:354(Orphanet)
- MONDO:0018149(MONDO)
- GARD:10891(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q5513690(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
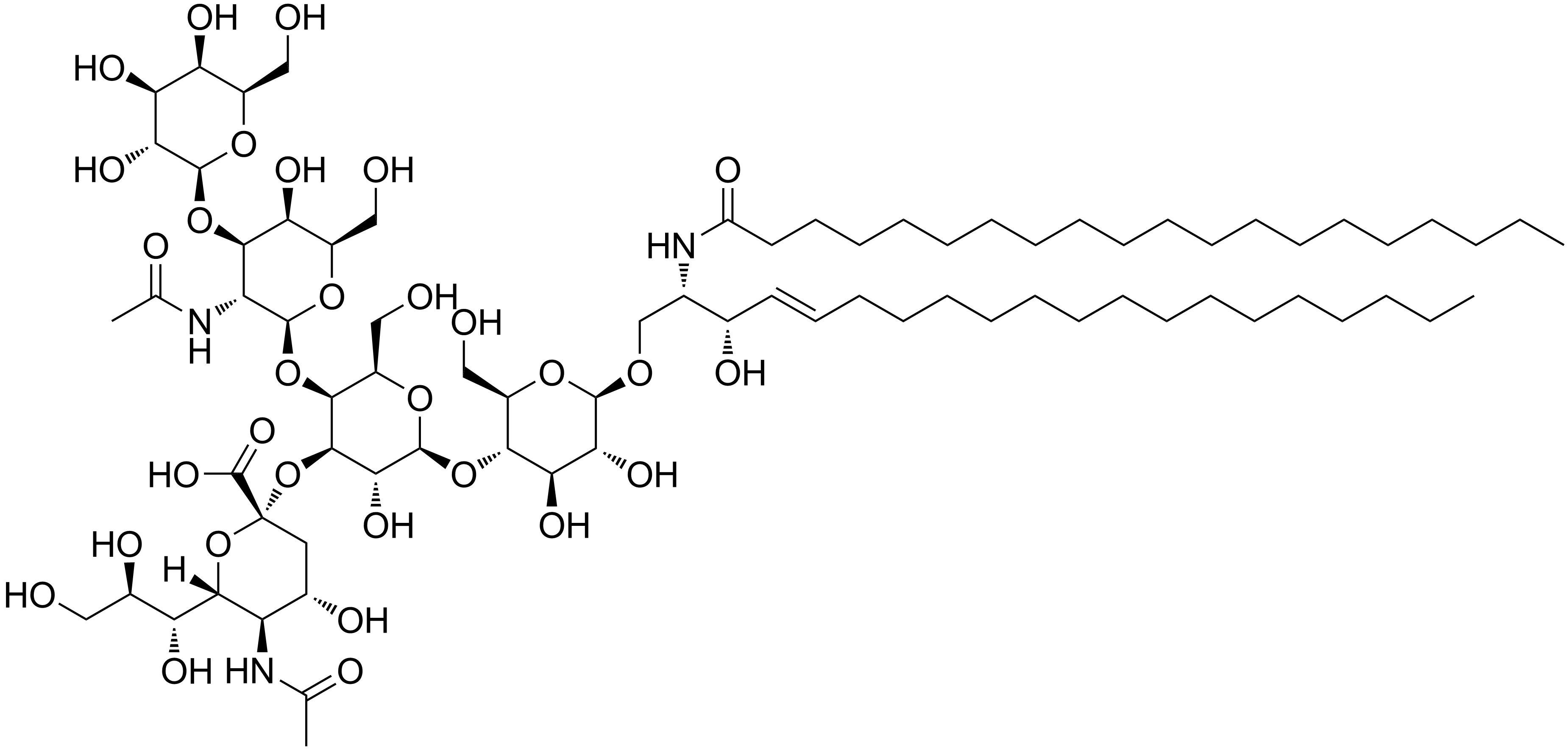
Gangliosidose GM1
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov