Displasia esquelética letal muito rara, caracterizada por hidropisia fetal, membros curtos e calcificação condro-óssea anormal. A doença é caracterizada por letalidade precoce no útero e os fetos afetados são considerados inviáveis.
Introdução
O que você precisa saber de cara
Displasia esquelética letal muito rara, caracterizada por hidropisia fetal, membros curtos e calcificação condro-óssea anormal. A doença é caracterizada por letalidade precoce no útero e os fetos afetados são considerados inviáveis.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 41 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 91 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisCatalyzes the reduction of the C14-unsaturated bond of lanosterol, as part of the metabolic pathway leading to cholesterol biosynthesis (PubMed:12618959, PubMed:16784888, PubMed:21327084, PubMed:27336722, PubMed:9630650). Plays a critical role in myeloid cell cholesterol biosynthesis which is essential to both myeloid cell growth and functional maturation (By similarity). Mediates the activation of NADPH oxidases, perhaps by maintaining critical levels of cholesterol required for membrane lipid
Nucleus inner membraneEndoplasmic reticulum membraneCytoplasmNucleus
Pelger-Huet anomaly
An autosomal dominant inherited abnormality of granulocytes, characterized by abnormal ovoid shape, reduced nuclear segmentation and an apparently looser chromatin structure.
Variantes genéticas (ClinVar)
87 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 106 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
11 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Displasia Greenberg
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
A Family of LBR Biallelic Pathogenic Variants Resulting in Rhizomelic Skeletal Dysplasia with Pelger-Huët Anomaly.
The lamin-B receptor (LBR) gene has two primary functions: maintaining the structural integrity of the nuclear envelope and playing a role in cholesterol biosynthesis. Heterozygous variants in the LBR gene have been associated with Pelger-Huët anomaly (PHA, OMIM #169400), while homozygous or compound heterozygous mutations have been associated with rhizomelic skeletal dysplasia, with or without PHA (OMIM #618019) and Greenberg dysplasia (OMIM #215140). We report a 4-year-old boy presenting with short stature and short limbs and his mother exhibiting milder findings. Genetic analysis revealed a heterozygous c.1640A>G (p.Asn547Ser) and c.43C>T (p.Arg15*) variants in the LBR gene in both the boy and his mother. The father was identified as a heterozygous carrier of the c.43C>T (p.Arg15*) variant. Peripheral blood smears confirmed the PHA in the patient and his parents. The phenotypic differences observed between the mother and male child in our study highlight the genetic variability and regressive nature of LBR-related skeletal dysplasias. This report shows the complexity of LBR-related phenotypes and expands the clinical spectrum of LBR mutations in rhizomelic skeletal dysplasia with PHA.
Novel LBR pathogenic variants with loss of sterol reductase activity participate in the pathogenesis of skeletal dysplasia via dysregulating canonical Wnt pathway.
Biallelic pathogenic variants in the lamin B receptor (LBR) with impaired sterol reductase function are associated with the development of perinatal lethal Greenberg dysplasia (GRBGD) and mild nonfatal skeletal dysplasia with or without Pelger-Huet anomaly (PHASK), as well as other related hereditary skeletal dysplasia. However, the underlying molecular mechanism remains unclear. In this study, we found two novel pathogenic variants of LBR, namely missense mutation (c.1011 T > G, NM_002296.4; p.Cys337Trp, NP_002287.2) and LBR gene deletion (Chr1q42.12 (225,515,082-225,633,464), NC_000001.10). LBR is a novel substrate of FBW7, which is degraded by GSK3β/FBW7-mediated proteasome pathway and whose C337W mutation promotes its degradation through enhanced interaction with FBW7. Wild-type but not C337W mutant LBR is upregulated by WNT3A-mediated inactivation of GSK3β/FBW7 axis and then participated in WNT3A-activated Wnt pathway through its mediated cholesterol synthesis. MC3T3-E1 cells with Lbr knockdown or cholesterol removal exhibited reduced mineralized nodules in the presence of WNT3A, but addition of cholesterol in the culture medium reversed this phenotype. Collectively, we detected two novel variants in LBR and our study revealed for the first time that disruption of cholesterol synthesis by LBR impairs Wnt pathway and thus disrupts the cell osteogenic differentiation, providing new insights into the pathogenesis of skeletal dysplasia caused by LBR variation.
Prenatal diagnosis of recurrent moderate skeletal dysplasias in lamin B receptors.
The lamin B receptor (LBR) gene is located in chromosome 1q42.12 and encodes the lamin B receptor, an intracellular protein that binds to lamin B. LBR mutations are associated with a broad phenotypic spectrum ranging from non-lethal to lethal skeletal dysplasias. The typical phenotypes include the Pelger-Huet anomaly (PHA) and embryonic lethal Greenberg dysplasia (GRBGD). With the further study of this gene, other phenotypes have been found in different individuals. This retrospective study analyzed recurrent prenatal moderate skeletal dysplasias in Chinese fetuses. Nothing malformed was detected in the fetal karyotype and microarray, while the whole-exome sequencing identified a homozygous variant (NM_002296.4:c.1757G>A, NP_002287.2:p.Arg586His) in exon 14 of the LBR gene in both fetuses. Mutation analysis in the parents confirmed that the c.1757G>A variation is heterozygous by Sanger sequencing. Intensive analysis on bioinformatics and familial co-segregation suggest that the homozygous variation in the LBR gene is responsible for this recurrent prenatal moderate skeletal dysplasia. Moreover, moderate skeletal dysplasias differ from typical GRBGD phenotypes. Our findings are based on the DNA base test and the prenatal diagnosis of skeletal dysplasia, which can be helpful in proper phenotyping and contribute to a better understanding of the correlation between the phenotype and genotype.
Antenatal diagnostic dilemma in a pseudodominant pedigree with lamin-B receptor (LBR)-related regressive spondylometaphyseal dysplasia.
The lamin-B receptor (LBR) encodes a dual-functioning inner nuclear membrane protein essential for cholesterol biosynthesis and chromatin organization. LBR pathogenic variants cause distinct phenotypes due to the dual function of LBR, including Pelger-Huët anomaly (PHA), PHA with mild skeletal anomalies (PHASK; MIM# 618019), LBR-related regressive type of spondylometaphyseal dysplasia (LBR-R-SMD), Greenberg dysplasia (MIM# 215140). We here report the first case with radiological manifestations of LBR-R-SMD in the fetal period, and milder skeletal findings in the similarly affected father. Direct sequencing of LBR revealed homozygous c.1534C>T (p.Arg512Trp) in exon 12 in both affected individuals. Our report further refines the early phenotype in LBR-R-SMD, and demonstrates that the p.Arg512Trp mutation is associated with PHA. We propose that LBR-R-SMD should be considered as a differential diagnosis in pregnancies with sonographic evidence of short and bowed tubular bones with narrow thorax. Evaluating peripheral blood smears of expectant parents for the presence of PHA may lead to a clinical diagnosis, allowing for comprehensive prenatal genetic counseling.
Deletion of LBR N-terminal domains recapitulates Pelger-Huet anomaly phenotypes in mouse without disrupting X chromosome inactivation.
Mutations in the gene encoding Lamin B receptor (LBR), a nuclear-membrane protein with sterol reductase activity, have been linked to rare human disorders. Phenotypes range from a benign blood disorder, such as Pelger-Huet anomaly (PHA), affecting the morphology and chromatin organization of white blood cells, to embryonic lethality as for Greenberg dysplasia (GRBGD). Existing PHA mouse models do not fully recapitulate the human phenotypes, hindering efforts to understand the molecular etiology of this disorder. Here we show, using CRISPR/Cas-9 gene editing technology, that a 236bp N-terminal deletion in the mouse Lbr gene, generating a protein missing the N-terminal domains of LBR, presents a superior model of human PHA. Further, we address recent reports of a link between Lbr and defects in X chromosome inactivation (XCI) and show that our mouse mutant displays minor X chromosome inactivation defects that do not lead to any overt phenotypes in vivo. We suggest that our N-terminal deletion model provides a valuable pre-clinical tool to the research community and will aid in further understanding the etiology of PHA and the diverse functions of LBR.
Publicações recentes
A Family of LBR Biallelic Pathogenic Variants Resulting in Rhizomelic Skeletal Dysplasia with Pelger-Huët Anomaly.
Novel LBR pathogenic variants with loss of sterol reductase activity participate in the pathogenesis of skeletal dysplasia via dysregulating canonical Wnt pathway.
Prenatal diagnosis of recurrent moderate skeletal dysplasias in lamin B receptors.
Antenatal diagnostic dilemma in a pseudodominant pedigree with lamin-B receptor (LBR)-related regressive spondylometaphyseal dysplasia.
Deletion of LBR N-terminal domains recapitulates Pelger-Huet anomaly phenotypes in mouse without disrupting X chromosome inactivation.
📚 EuropePMC8 artigos no totalmostrando 10
A Family of LBR Biallelic Pathogenic Variants Resulting in Rhizomelic Skeletal Dysplasia with Pelger-Huët Anomaly.
Molecular syndromologyNovel LBR pathogenic variants with loss of sterol reductase activity participate in the pathogenesis of skeletal dysplasia via dysregulating canonical Wnt pathway.
Biochimica et biophysica acta. Molecular basis of diseasePrenatal diagnosis of recurrent moderate skeletal dysplasias in lamin B receptors.
Frontiers in geneticsAntenatal diagnostic dilemma in a pseudodominant pedigree with lamin-B receptor (LBR)-related regressive spondylometaphyseal dysplasia.
American journal of medical genetics. Part ADeletion of LBR N-terminal domains recapitulates Pelger-Huet anomaly phenotypes in mouse without disrupting X chromosome inactivation.
Communications biologyA homozygous variant in the Lamin B receptor gene LBR results in a non-lethal skeletal dysplasia without Pelger-Huët anomaly.
BoneA new case of Greenberg dysplasia and literature review suggest that Greenberg dysplasia, dappled diaphyseal dysplasia, and Astley-Kendall dysplasia are allelic disorders.
Molecular genetics & genomic medicineA novel case of Greenberg dysplasia and genotype-phenotype correlation analysis for LBR pathogenic variants: An instructive example of one gene-multiple phenotypes.
American journal of medical genetics. Part ALamin B receptor-related disorder is associated with a spectrum of skeletal dysplasia phenotypes.
BoneRadiographic features of the skeleton in disorders of post-squalene cholesterol biosynthesis.
Pediatric radiologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Displasia Greenberg.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Displasia Greenberg
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- A Family of LBR Biallelic Pathogenic Variants Resulting in Rhizomelic Skeletal Dysplasia with Pelger-Huët Anomaly.
- Novel LBR pathogenic variants with loss of sterol reductase activity participate in the pathogenesis of skeletal dysplasia via dysregulating canonical Wnt pathway.
- Prenatal diagnosis of recurrent moderate skeletal dysplasias in lamin B receptors.
- Antenatal diagnostic dilemma in a pseudodominant pedigree with lamin-B receptor (LBR)-related regressive spondylometaphyseal dysplasia.
- Deletion of LBR N-terminal domains recapitulates Pelger-Huet anomaly phenotypes in mouse without disrupting X chromosome inactivation.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1426(Orphanet)
- OMIM OMIM:215140(OMIM)
- MONDO:0008974(MONDO)
- GARD:8754(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q3335662(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
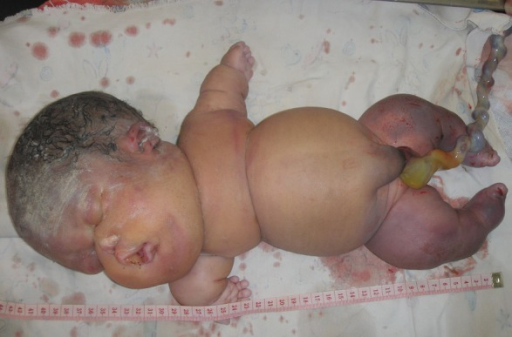
Displasia Greenberg
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata