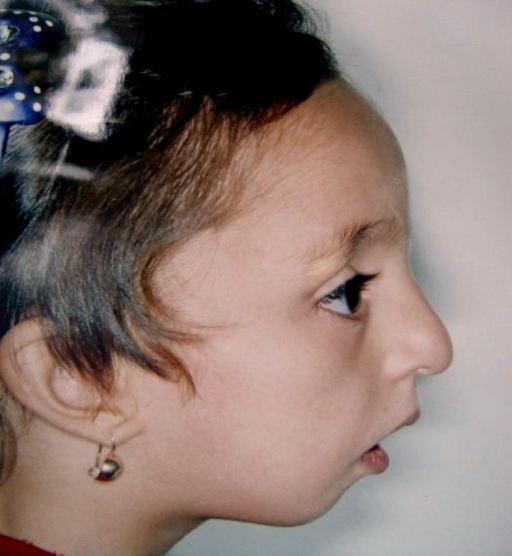
A Disostose Acrofacial, tipo Catania, é uma condição muito rara caracterizada por: * Crescimento lento do bebê ainda no útero. * Baixa estatura após o nascimento. * Microcefalia (cabeça menor que o normal). * Linha do cabelo em forma de "V" na testa (chamado "pico de viúva"). * Malformações no rosto e mandíbula, mas sem fenda no céu da boca. * Cáries frequentes. * Desenvolvimento levemente incompleto dos braços e pernas, com dedos das mãos e pés curtos (braquidactilia). * Uma leve união entre os dedos, formando uma pequena membrana. * Uma única linha na palma da mão (prega simiesca). * Hérnia inguinal. * Em meninos: testículos que não desceram (criptorquidia) e hipospadia (abertura da uretra em um local anormal no pênis).
Introdução
O que você precisa saber de cara
A Disostose Acrofacial, tipo Catania, é uma condição muito rara caracterizada por: * Crescimento lento do bebê ainda no útero. * Baixa estatura após o nascimento. * Microcefalia (cabeça menor que o normal). * Linha do cabelo em forma de "V" na testa (chamado "pico de viúva"). * Malformações no rosto e mandíbula, mas sem fenda no céu da boca. * Cáries frequentes. * Desenvolvimento levemente incompleto dos braços e pernas, com dedos das mãos e pés curtos (braquidactilia). * Uma leve união entre os dedos, formando uma pequena membrana. * Uma única linha na palma da mão (prega simiesca). * Hérnia inguinal. * Em meninos: testículos que não desceram (criptorquidia) e hipospadia (abertura da uretra em um local anormal no pênis).
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 16 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 47 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Disostose acrofacial, tipo Catania
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Publicações recentes
A novel EVC2 splice-site variant expands the mutational and phenotypic spectrum of Weyers acrofacial dysostosis.
A New Case of Nager Syndrome as a Rare Cause of Acrofacial Dysostosis.
The Phenotypic Spectrum of Miller Syndrome: Insight From a French Cohort.
RNA Polymerase I Dysfunction Underlying Craniofacial Syndromes: Integrated Genetic Analysis Reveals Parallels to 22q11.2 Deletion Syndrome.
Facial Bone Defects Associated with Lateral Facial Clefts Tessier Type 6, 7 and 8 in Syndromic Neurocristopathies: A Detailed Micro-CT Analysis on Historical Museum Specimens.
Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Disostose acrofacial, tipo Catania.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Disostose acrofacial, tipo Catania
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Broadening the spectrum of Catania brachydactylous type of acrofacial dysostoses.
- A novel EVC2 splice-site variant expands the mutational and phenotypic spectrum of Weyers acrofacial dysostosis.
- A New Case of Nager Syndrome as a Rare Cause of Acrofacial Dysostosis.
- The Phenotypic Spectrum of Miller Syndrome: Insight From a French Cohort.
- RNA Polymerase I Dysfunction Underlying Craniofacial Syndromes: Integrated Genetic Analysis Reveals Parallels to 22q11.2 Deletion Syndrome.
- Facial Bone Defects Associated with Lateral Facial Clefts Tessier Type 6, 7 and 8 in Syndromic Neurocristopathies: A Detailed Micro-CT Analysis on Historical Museum Specimens.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1786(Orphanet)
- OMIM OMIM:101805(OMIM)
- MONDO:0007045(MONDO)
- GARD:494(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Q21154052(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Disostose acrofacial, tipo Catania
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata