A sialúria é um distúrbio metabólico extremamente raro, descrito em menos de 10 pacientes até o momento e caracterizado por sinais e sintomas variáveis, principalmente na infância, incluindo retardo de crescimento transitório, icterícia neonatal levemente prolongada, hepatomegalia equívoca ou leve, anemia microcítica, infecções respiratórias superiores frequentes, gastroenterite, desidratação e fácies plana e grosseira. Dificuldades de aprendizagem e convulsões podem ocorrer na infância.
Introdução
O que você precisa saber de cara
A sialúria é um distúrbio metabólico extremamente raro, descrito em menos de 10 pacientes até o momento e caracterizado por sinais e sintomas variáveis, principalmente na infância, incluindo retardo de crescimento transitório, icterícia neonatal levemente prolongada, hepatomegalia equívoca ou leve, anemia microcítica, infecções respiratórias superiores frequentes, gastroenterite, desidratação e fácies plana e grosseira. Dificuldades de aprendizagem e convulsões podem ocorrer na infância.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 20 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 56 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisBifunctional enzyme that possesses both UDP-N-acetylglucosamine 2-epimerase and N-acetylmannosamine kinase activities, and serves as the initiator of the biosynthetic pathway leading to the production of N-acetylneuraminic acid (NeuAc), a critical precursor in the synthesis of sialic acids. By catalyzing this pivotal and rate-limiting step in sialic acid biosynthesis, this enzyme assumes a pivotal role in governing the regulation of cell surface sialylation, playing a role in embryonic angiogene
Cytoplasm, cytosol
Sialuria
In sialuria, free sialic acid accumulates in the cytoplasm and gram quantities of neuraminic acid are secreted in the urine. The metabolic defect involves lack of feedback inhibition of UDP-GlcNAc 2-epimerase by CMP-Neu5Ac, resulting in constitutive overproduction of free Neu5Ac. Clinical features include variable degrees of developmental delay, coarse facial features and hepatomegaly. Sialuria inheritance is autosomal dominant.
Variantes genéticas (ClinVar)
389 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 916 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Sialúria
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Disorders in sialic acid metabolism and sialylation pathway.
Sialic acids (Sias) are acidic 9‑carbon monosaccharides. Both free Sias and conjugated Sias (sialylation) exist in the human body and have decisive impacts on human health and disease. Cellular free Sias are made via de novo biosynthesis, recycled from lysosomal salvage, and even by uptake of extracellular Sias, respectively. Sialylation of glycoproteins and glycolipids are catalyzed by sialyltransferases using CMP-Sia as the donor in the Golgi apparatus. In addition, free Sia can be degraded/catabolized into ManNAc and pyruvate in the cytosol. Overall, cellular free Sia and sialylation are kept at certain levels for normal cell functions. However, Sias deficiency and overproduction (accumulation), hyposialylation (undersialylation) and hypersialylation all cause disorders in the human body through a variety of mechanisms, but most of them are still not fully clarified. This review discusses recent understanding of disorders in Sia biosynthesis, salvage, catabolism, and sialylation pathways and therapeutic explorations for these disorders as well.
Expression of GNE mutant proteins increases CHO intracellular CMP-Neu5Ac levels without impact on bioprocess performance.
Modulation of various nucleotide sugar levels in cells has been demonstrated as an effective way to alter the composition of N-glycans. Previous studies have demonstrated the ability to impact CMP-Neu5Ac levels by the addition of N-acetylated mannosamine (ManNAc) to culture media. In this study, the relationship between adding varying levels of ManNAc to cell cultures and the impact on both CMP-Neu5Ac levels and cell growth were examined. Increasing the concentration of ManNAc added resulted in higher levels of CMP-Neu5Ac, but negatively impacted cell growth. Through cellular genetic engineering, we sought to devise an alternative method of increasing ManNAc levels without impacting cell growth. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine-kinase (GNE) gene is the rate-limiting enzyme in which congenital mutations can cause Sialuria, a rare metabolic disorder characterized by cytoplasmic accumulation and urinary excretion of free sialic acid. A mutant form of the GNE gene, harboring three mutations (D53H, R263I, R266Q), was site-specifically integrated (SSI) into one locus in CHO cells. This mutant protein dramatically increased the intracellular concentrations of CMP-Neu5Ac, reaching the maximal level as with the addition of ManNAc. These data together indicate that the GNE mutants could provide an effective way for substituting the high-cost supplementation of ManNAc without impacting cell growth. The investigation has also demonstrated the feasibility of the dual-landing-pad SSI cell line engineering approach for improving product qualities of biotherapeutics.
Psychiatric symptoms in Salla disease.
Salla disease (SD) is a rare lysosomal storage disorder characterised by intellectual disability ataxia, athetosis, nystagmus, and central nervous system demyelination. Although the neurological spectrum of SD's clinical phenotype is well defined, psychotic symptoms in SD remain unreported. We reviewed the presence of psychiatric symptoms in patients diagnosed with SD. Medical records of all SD patients at Oulu University Hospital during the years 1982-2015 were systematically reviewed to evaluate the presence of psychiatric symptoms. Psychiatric symptoms were frequently associated with SD (10/24, 42%), and two patients were described as developing psychosis as adolescents. We reported their clinical characteristics in detail and assessed the prevalence of psychiatric symptoms in a cohort of 24 patients. Other psychiatric factors associated with SD were sleeping disorders (8/24, 32%), aggressive behaviour disorders or restlessness (6/24, 25%), and off-label antipsychotic medication (4/24, 17%). This report expands the knowledge of the phenotypic spectrum of SD and demonstrates the importance of recognising the possibility of psychiatric symptoms, including psychosis, in persons with SD.
Free urinary sialic acid levels may be elevated in patients with pneumococcal sepsis.
Urine free sialic acid (UFSA) is an important diagnostic biomarker for sialuria (GNE variants) and infantile sialic acid storage disease/Salla disease (SLC17A5 variants). Traditionally, UFSA has been measured using specific single-plex methodology in relatively small cohorts of patients with clinical symptoms suggestive of these disorders. The use of multiplex tandem mass spectrometry urine screening (UMSMS) has meant that UFSA can be measured semi-quantitatively in a much larger cohort of patients being investigated for suspected metabolic disorders. We hypothesised that the neuraminidase of Streptococcus pneumoniae may release free sialic acid from endogenous sialylated glycoconjugates and result in increased UFSA levels. We conducted a retrospective review of clinical records of patients who were identified as having S. pneumoniae infection and who also had UMSMS at the time of their acute infection. We identified three cases of increased UFSA detected by UMSMS screening that were secondary to S. pneumoniae sepsis. Additional testing ruled out genetic causes of increased UFSA in the first patient. All three patients had overwhelming sepsis with multiorgan dysfunction which was fatal. Glycosylation abnormalities consistent with the removal of sialic acid were demonstrated in serum transferrin patterns in one patient. We have demonstrated in a retrospective cohort that elevation of UFSA levels have been observed in cases of S. pneumoniae sepsis. This expands our knowledge of UFSA as a biomarker in human disease. This research demonstrates that infection with organisms with neuraminidase activity should be considered in patients with unexplained increases in UFSA.
Recombinant human N-acetylneuraminate lyase as a tool to study clinically relevant mutant variants.
N-acetylneuraminic acid (sialic acid) is an abundantly found carbohydrate moiety covering the surface of all vertebrate cells and secreted glycoproteins. The human N-acetylneuraminate pyruvate lyase (NPL) interconverts sialic acid to N-acetylmannosamine and pyruvate, and mutations of the NPL gene were found to cause sialuria and impair the functionality of muscles. Here we report the soluble and functional expression of human NPL in Escherichia coli, which allowed us to study the biochemical properties of two clinically relevant NLP mutations (Asn45Asp and Arg63Cys). The Asn45Asp mutant variant was enzymatically active, but had lower expression levels and showed reduced stability when compared to the wild-type NPL variant. Expression trials of the Arg63Cys mutant did not yield any recombinant protein and consequently, no enzymatic activity was detected. The locations of these clinically relevant amino acid substitutions are also discussed by using a human NPL homology model.
Publicações recentes
Disorders in sialic acid metabolism and sialylation pathway.
Expression of GNE mutant proteins increases CHO intracellular CMP-Neu5Ac levels without impact on bioprocess performance.
Free urinary sialic acid levels may be elevated in patients with pneumococcal sepsis.
Psychiatric symptoms in Salla disease.
Recombinant human N-acetylneuraminate lyase as a tool to study clinically relevant mutant variants.
📚 EuropePMC38 artigos no totalmostrando 14
Disorders in sialic acid metabolism and sialylation pathway.
Life sciencesExpression of GNE mutant proteins increases CHO intracellular CMP-Neu5Ac levels without impact on bioprocess performance.
Bioprocess and biosystems engineeringFree urinary sialic acid levels may be elevated in patients with pneumococcal sepsis.
Clinical chemistry and laboratory medicinePsychiatric symptoms in Salla disease.
European child & adolescent psychiatryRecombinant human N-acetylneuraminate lyase as a tool to study clinically relevant mutant variants.
Carbohydrate researchNovel GNE Gene Variants Associated with Severe Congenital Thrombocytopenia and Platelet Sialylation Defect.
Thrombosis and haemostasisSialuria-Related Intellectual Disability in Children and Adolescent of Pakistan: Tenth Patient Described has a Novel Mutation in the GNE Gene.
CNS & neurological disorders drug targetsSialic acid catabolism by N-acetylneuraminate pyruvate lyase is essential for muscle function.
JCI insightSialuria: Ninth Patient Described Has a Novel Mutation in GNE.
JIMD reportsRole of IGF-1R in ameliorating apoptosis of GNE deficient cells.
Scientific reportsIncreased Polysialylation of the Neural Cell Adhesion Molecule in a Transgenic Mouse Model of Sialuria.
Chembiochem : a European journal of chemical biologyNew observation of sialuria prompts detection of liver tumor in previously reported patient.
Molecular genetics and metabolismMechanism and inhibition of human UDP-GlcNAc 2-epimerase, the key enzyme in sialic acid biosynthesis.
Scientific reportsEngineering Sialic Acid Synthesis Ability in Insect Cells.
Methods in molecular biology (Clifton, N.J.)Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Sialúria.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Sialúria
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Disorders in sialic acid metabolism and sialylation pathway.
- Expression of GNE mutant proteins increases CHO intracellular CMP-Neu5Ac levels without impact on bioprocess performance.
- Psychiatric symptoms in Salla disease.
- Free urinary sialic acid levels may be elevated in patients with pneumococcal sepsis.
- Recombinant human N-acetylneuraminate lyase as a tool to study clinically relevant mutant variants.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:3166(Orphanet)
- OMIM OMIM:269921(OMIM)
- MONDO:0010028(MONDO)
- GARD:4865(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q7506696(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
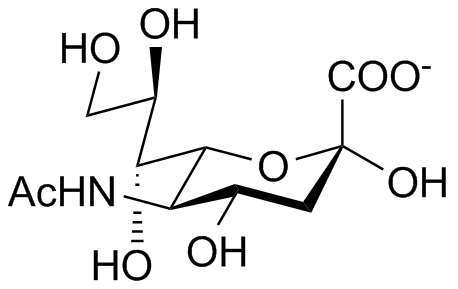
Sialúria
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata