Doença genética rara autossômica dominante, com início na infância, caracterizada por episódios de dor intensa, predominantemente nos membros superiores. A condição está associada a mutações no gene TRPA1.
Introdução
O que você precisa saber de cara
Visão geral
A Síndrome de dor familiar episódica com envolvimento predominante dos membros superiores é uma doença genética rara, caracterizada por episódios de dor intensa que afetam principalmente a parte superior do corpo. A condição tem início precoce, ainda na infância ou mesmo no período neonatal, e segue um padrão de herança autossômica dominante — ou seja, uma cópia alterada do gene responsável já é suficiente para causar a doença. A prevalência estimada é de menos de 1 caso por 1.000.000 de pessoas, sendo considerada uma condição extremamente rara.[1][4]
Sinais e sintomas
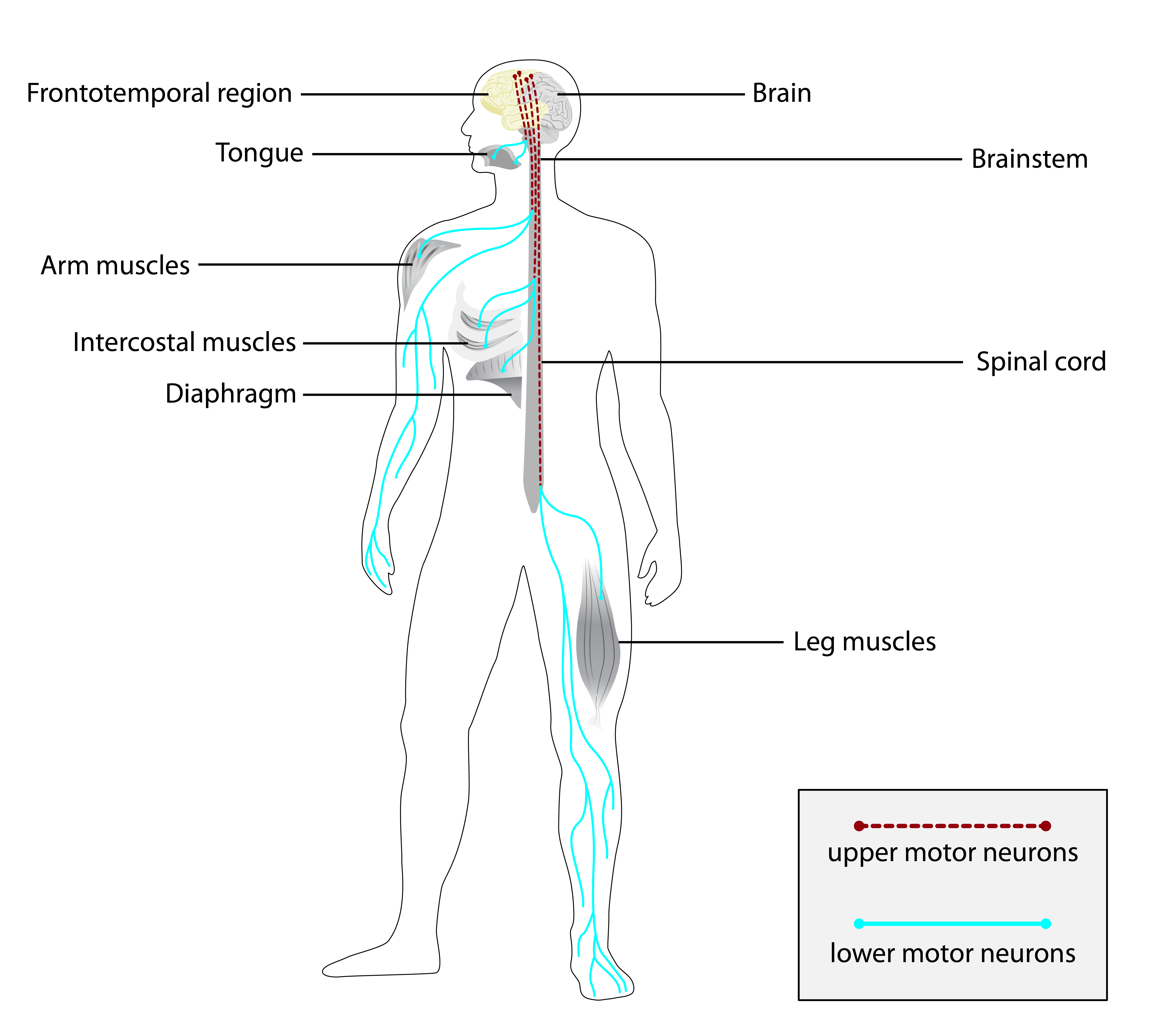
Os principais sintomas são episódios de dor que começam na infância, sem um gatilho aparente, e que afetam predominantemente os membros superiores (braços, ombros e tórax). A dor é descrita como intensa e pode durar minutos a horas, com intervalos variáveis entre os episódios. Durante as crises, a pessoa pode apresentar sudorese, palidez e sensação de desconforto generalizado. Fora dos episódios, não há dor ou outros sintomas.[1][4]
Causas genéticas
A síndrome é causada por variantes patogênicas no gene TRPA1 (Transient Receptor Potential Cation Channel Subfamily A Member 1). Esse gene fornece instruções para a produção de um canal iônico que atua na percepção da dor e na resposta a estímulos químicos e térmicos. A herança é autossômica dominante, o que significa que filhos de uma pessoa afetada têm 50% de chance de herdar a condição.[1][2][5]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos episódios de dor e na história familiar. A confirmação é feita por meio de teste genético molecular, que identifica variantes patogênicas no gene TRPA1. Atualmente, há 33 variantes reportadas no ClinVar e 7 testes genéticos disponíveis para essa condição. O código CID-10 associado é M79.6 (Dor não especificada nos membros).[1][2][5]
Tratamento e manejo
Não há um tratamento específico aprovado para a síndrome. O manejo é sintomático e focado no alívio da dor durante os episódios. Medicamentos analgésicos comuns podem ser utilizados, mas a resposta varia entre os pacientes. É importante que o acompanhamento seja feito por uma equipe multidisciplinar, incluindo geneticista e especialista em dor. No Brasil, a condição não possui cobertura pelo SUS para tratamentos específicos.[1][2]
Prognóstico e qualidade de vida
O prognóstico é variável. Os episódios de dor podem ser debilitantes durante a infância, mas algumas pessoas relatam melhora com o avançar da idade. Não há evidências de que a síndrome reduza a expectativa de vida. O suporte psicológico e o manejo adequado da dor podem melhorar significativamente a qualidade de vida dos pacientes e de suas famílias.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Doença genética rara autossômica dominante, com início na infância, caracterizada por episódios de dor intensa, predominantemente nos membros superiores. A condição está associada a mutações no gene TRPA1.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Síndrome de dor familiar episódica com envolvimento predominante dos membros superiores é uma doença genética rara, caracterizada por episódios de dor intensa que afetam principalmente a parte superior do corpo. A condição tem início precoce, ainda na infância ou mesmo no período neonatal, e segue um padrão de herança autossômica dominante — ou seja, uma cópia alterada do gene responsável já é suficiente para causar a doença. A prevalência estimada é de menos de 1 caso por 1.000.000 de pessoas, sendo considerada uma condição extremamente rara.[1][4]
Sinais e sintomas
Os principais sintomas são episódios de dor que começam na infância, sem um gatilho aparente, e que afetam predominantemente os membros superiores (braços, ombros e tórax). A dor é descrita como intensa e pode durar minutos a horas, com intervalos variáveis entre os episódios. Durante as crises, a pessoa pode apresentar sudorese, palidez e sensação de desconforto generalizado. Fora dos episódios, não há dor ou outros sintomas.[1][4]
Causas genéticas
A síndrome é causada por variantes patogênicas no gene TRPA1 (Transient Receptor Potential Cation Channel Subfamily A Member 1). Esse gene fornece instruções para a produção de um canal iônico que atua na percepção da dor e na resposta a estímulos químicos e térmicos. A herança é autossômica dominante, o que significa que filhos de uma pessoa afetada têm 50% de chance de herdar a condição.[1][2][5]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos episódios de dor e na história familiar. A confirmação é feita por meio de teste genético molecular, que identifica variantes patogênicas no gene TRPA1. Atualmente, há 33 variantes reportadas no ClinVar e 7 testes genéticos disponíveis para essa condição. O código CID-10 associado é M79.6 (Dor não especificada nos membros).[1][2][5]
Tratamento e manejo
Não há um tratamento específico aprovado para a síndrome. O manejo é sintomático e focado no alívio da dor durante os episódios. Medicamentos analgésicos comuns podem ser utilizados, mas a resposta varia entre os pacientes. É importante que o acompanhamento seja feito por uma equipe multidisciplinar, incluindo geneticista e especialista em dor. No Brasil, a condição não possui cobertura pelo SUS para tratamentos específicos.[1][2]
Prognóstico e qualidade de vida
O prognóstico é variável. Os episódios de dor podem ser debilitantes durante a infância, mas algumas pessoas relatam melhora com o avançar da idade. Não há evidências de que a síndrome reduza a expectativa de vida. O suporte psicológico e o manejo adequado da dor podem melhorar significativamente a qualidade de vida dos pacientes e de suas famílias.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 3 características clínicas mais associadas, ordenadas por frequência.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Ligand-activated Ca(2+)-permeable, nonselective cation channel involved in pain detection and possibly also in cold perception, oxygen concentration perception, cough, itch, and inner ear function (PubMed:17259981, PubMed:21195050, PubMed:21873995, PubMed:23199233, PubMed:25389312, PubMed:33152265). Has a relatively high Ca(2+) selectivity, with a preference for divalent over monovalent cations (Ca(2+) > Ba(2+) > Mg(2+) > NH4(+) > Li(+) > K(+)), the influx of cation into the cytoplasm leads to m
Cell membrane
Episodic pain syndrome, familial, 1
An autosomal dominant neurologic disorder characterized by onset in infancy of episodic debilitating upper body pain triggered by fasting, cold, and physical stress. The period of intense pain is accompanied by breathing difficulties, tachycardia, sweating, generalized pallor, peribuccal cyanosis, and stiffness of the abdominal wall. Affected individuals do not manifest altered pain sensitivity outside the episodes.
Variantes genéticas (ClinVar)
33 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 23 variantes classificadas pelo ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de dor familiar episódica com envolvimento predominante dos membros superiores
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de dor familiar episódica com envolvimento predominante dos membros superiores.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de dor familiar episódica com envolvimento predominante dos membros superiores
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:391389(Orphanet)
- OMIM OMIM:615040(OMIM)
- MONDO:0014021(MONDO)
- GARD:17618(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Q55784462(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome de dor familiar episódica com envolvimento predominante dos membros superiores
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata