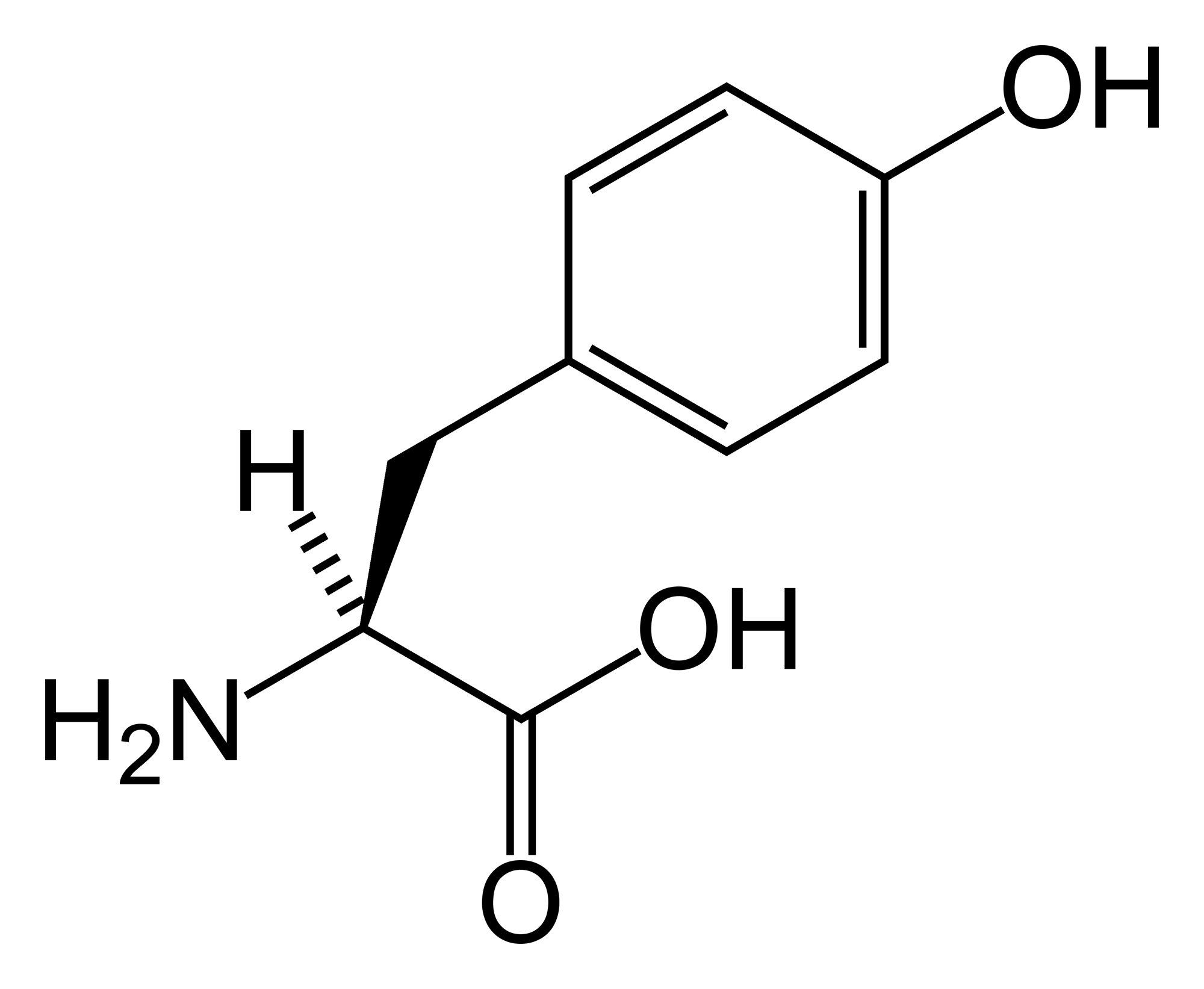
A tirosinemia tipo 3 é um erro congênito do metabolismo da tirosina caracterizado por hipertirosinemia leve e aumento da excreção urinária de 4-hidroxifenilpiruvato, 4-hidroxifenillactato e 4-hidroxifenilacetato.
Introdução
O que você precisa saber de cara
A tirosinemia tipo 3 é um erro congênito do metabolismo da tirosina caracterizado por hipertirosinemia leve e aumento da excreção urinária de 4-hidroxifenilpiruvato, 4-hidroxifenillactato e 4-hidroxifenilacetato.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 6 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 11 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisCatalyzes the conversion of 4-hydroxyphenylpyruvic acid to homogentisic acid, one of the steps in tyrosine catabolism
CytoplasmEndoplasmic reticulum membraneGolgi apparatus membrane
Tyrosinemia 3
An autosomal recessive inborn error of metabolism characterized by high levels of tyrosine in the blood, massive excretion of its derivatives into urine, seizures and mild intellectual disability.
Medicamentos aprovados (FDA)
1 medicamento encontrado nos registros da FDA americana.
Variantes genéticas (ClinVar)
91 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 1.607 variantes classificadas pelo ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Tirosinemia tipo III
Centros de Referência SUS
21 centros habilitados pelo SUS para Tirosinemia tipo III
Centros para Tirosinemia tipo III
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
NUPAD / Faculdade de Medicina UFMG
Av. Prof. Alfredo Balena, 189 - 5 andar - Centro, Belo Horizonte - MG, 30130-100 · CNES 2183226
Serviço de Referência
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital de Clínicas da Universidade Federal de Pernambuco
Av. Prof. Moraes Rego, 1235 - Cidade Universitária, Recife - PE, 50670-901 · CNES 2561492
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital Universitário Onofre Lopes (HUOL)
Av. Nilo Peçanha, 620 - Petrópolis, Natal - RN, 59012-300 · CNES 2408570
Atenção Especializada
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
Instituto da Criança e do Adolescente (ICr-HCFMUSP)
Av. Dr. Enéas Carvalho de Aguiar, 647 - Cerqueira César, São Paulo - SP, 05403-000 · CNES 2081695
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
2 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
21 ensaios clínicos encontrados, 3 ativos.
Publicações mais relevantes
Different Clinic, Different Diagnosis: Tyrosinemia Type 3.
Tyrosinemia type III is an extremely rare autosomal recessive metabolic disorder resulting from biallelic pathogenic mutations in the HPD gene. To date, only a limited number of cases have been reported worldwide, and the full spectrum of clinical manifestations remains incompletely understood. While neurodevelopmental abnormalities are the most commonly described features, ocular involvement has rarely been documented. Here, we present a 9-month-old girl who exhibited strikingly atypical ocular symptoms characterized by persistent severe photophobia and allergic conjunctivitis at initial presentation. This combination of findings has been reported only sporadically in the literature. Biochemical and genetic investigations confirmed the diagnosis by identifying two novel heterozygous variants in the HPD gene. Implementation of a phenylalanine- and tyrosine-restricted diet led to a marked reduction in plasma tyrosine concentrations and improvement in clinical symptoms. This case underscores the diagnostic importance of considering inherited metabolic disorders in infants presenting with severe photosensitivity and conjunctival irritation, even in the absence of other systemic features. Furthermore, it highlights the need for long-term follow-up to monitor potential neurological and ocular complications associated with persistently elevated tyrosine levels.
Coexistence of phenylketonuria and tyrosinemia type 3: challenges in the dietary management.
Phenylketonuria (PKU) and tyrosinemia type 3 (HT3) are both rare autosomal recessive disorders of phenylalanine-tyrosine metabolism. PKU is caused by a deficiency in phenylalanine hydroxylase (PAH), leading to elevated phenylalanine (Phe) and reduced tyrosine (Tyr) levels. HT3, the rarest form of tyrosinemia, is due to a deficiency in 4-hydroxyphenylpyruvate dioxygenase (HPD). We report a 5-year-old girl diagnosed with both PKU and HT3. She presented with elevated Phe levels in neonatal screening, and subsequent biochemical tests revealed both hyperphenylalaninemia and elevated Tyr levels. Genetic analysis confirmed the diagnoses, identifying homozygous mutations in both the PAH and HPD genes. Dietary management to maintain optimal Phe and Tyr levels proved to be challenging due to the presence of these two coexisting pathologies especially during infections and due to dietary non-compliance, necessitating frequent adjustments in the treatment strategy. This case highlights the importance of considering multiple metabolic disorders in patients with unexplained clinical and biochemical findings. Early diagnosis and stringent dietary management are crucial for preventing neurological damage and ensuring favorable outcomes in patients with concurrent metabolic disorders.
A Novel Genetic Screen Identifies Modifiers of Age-Dependent Amyloid β Toxicity in the Drosophila Brain.
The accumulation of amyloid β peptide (Aβ) in the brain of Alzheimer's disease (AD) patients begins many years before clinical onset. Such process has been proposed to be pathogenic through the toxicity of Aβ soluble oligomers leading to synaptic dysfunction, phospho-tau aggregation and neuronal loss. Yet, a massive accumulation of Aβ can be found in approximately 30% of aged individuals with preserved cognitive function. Therefore, within the frame of the "amyloid hypothesis", compensatory mechanisms and/or additional neurotoxic or protective factors need to be considered and investigated. Here we describe a modifier genetic screen in Drosophila designed to identify genes that modulate toxicity of Aβ42 in the CNS. The expression of Aβ42 led to its accumulation in the brain and a moderate impairment of negative geotaxis at 18 days post-eclosion (d.p.e) as compared with genetic or parental controls. These flies were mated with a collection of lines carrying chromosomal deletions and negative geotaxis was assessed at 5 and 18 d.p.e. Our screen is the first to take into account all of the following features, relevant to sporadic AD: (1) pan-neuronal expression of wild-type Aβ42; (2) a quantifiable complex behavior; (3) Aβ neurotoxicity associated with progressive accumulation of the peptide; and (4) improvement or worsening of climbing ability only evident in aged animals. One hundred and ninety-nine deficiency (Df) lines accounting for ~6300 genes were analyzed. Six lines, including the deletion of 52 Drosophila genes with human orthologs, significantly modified Aβ42 neurotoxicity in 18-day-old flies. So far, we have validated CG11796 and identified CG17249 as a strong candidate (whose human orthologs are HPD and PRCC, respectively) by using RNAi or mutant hemizygous lines. PRCC encodes proline-rich protein PRCC (ppPRCC) of unknown function associated with papillary renal cell carcinoma. HPD encodes 4-hydroxyphenylpyruvate dioxygenase (HPPD), a key enzyme in tyrosine degradation whose Df causes autosomal recessive Tyrosinemia type 3, characterized by mental retardation. Interestingly, lines with a partial Df of HPD ortholog showed increased intraneuronal accumulation of Aβ42 that coincided with geotaxis impairment. These previously undetected modifiers of Aβ42 neurotoxicity in Drosophila warrant further study to validate their possible role and significance in the pathogenesis of sporadic AD.
Tyrosinemia type III in an asymptomatic girl.
Tyrosinemia type 3 (HT3) is a rare inborn error of tyrosine metabolism caused by mutations in the HPD gene encoding 4-hydroxyphenyl-pyruvate dioxygenase, which is transmitted in an autosomal recessive trait. The disorder is characterized by tyrosine accumulation in body fluids and massive excretion of tyrosine derivatives into urine (www.orpha.net). Since it is the least frequent form of tyrosinemia, only few cases with the variable but rather mild clinical features have been described so far. We report an 11 year old girl presenting with no clinical symptoms and with normal mental development who has been diagnosed with HT3 through metabolic screening on the basis of elevated serum level of tyrosine ranging from 425 to 535 μmol/L (normal values: 29-86 μmol/L), and elevated urinary excretion of p-hydroxyphenyl derivatives confirmed genetically with the homozygous c.479A > G (p.Tyr160Cys) missense change in the HPD gene. The girl has been only presenting with recurrent proteinuria of unknown etiology. A phenylalanine- and tyrosine-restricted diet has never been administered. Presented case may suggest that high tyrosine concentration itself does not participate directly in neuronal damage described in patients with tyrosinemia type 3.
Publicações recentes
Different Clinic, Different Diagnosis: Tyrosinemia Type 3.
Coexistence of phenylketonuria and tyrosinemia type 3: challenges in the dietary management.
A Novel Genetic Screen Identifies Modifiers of Age-Dependent Amyloid β Toxicity in the Drosophila Brain.
Tyrosinemia type III in an asymptomatic girl.
Fungal metabolic model for tyrosinemia type 3: molecular characterization of a gene encoding a 4-hydroxy-phenyl pyruvate dioxygenase from Aspergillus nidulans.
📚 EuropePMC697 artigos no totalmostrando 4
Different Clinic, Different Diagnosis: Tyrosinemia Type 3.
Molecular syndromologyCoexistence of phenylketonuria and tyrosinemia type 3: challenges in the dietary management.
Journal of pediatric endocrinology & metabolism : JPEMA Novel Genetic Screen Identifies Modifiers of Age-Dependent Amyloid β Toxicity in the Drosophila Brain.
Frontiers in aging neuroscienceTyrosinemia type III in an asymptomatic girl.
Molecular genetics and metabolism reportsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Tirosinemia tipo III.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Tirosinemia tipo III
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Different Clinic, Different Diagnosis: Tyrosinemia Type 3.
- Coexistence of phenylketonuria and tyrosinemia type 3: challenges in the dietary management.
- A Novel Genetic Screen Identifies Modifiers of Age-Dependent Amyloid β Toxicity in the Drosophila Brain.
- Tyrosinemia type III in an asymptomatic girl.
- Fungal metabolic model for tyrosinemia type 3: molecular characterization of a gene encoding a 4-hydroxy-phenyl pyruvate dioxygenase from Aspergillus nidulans.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:69723(Orphanet)
- OMIM OMIM:276710(OMIM)
- MONDO:0010162(MONDO)
- GARD:10332(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q7861692(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Tirosinemia tipo III
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos aprovados FDA
- fonte: FDA OpenFDA
- Reposicionamento
- fonte: Drug Repurposing Hub
- Ensaios clínicos
- fonte: ClinicalTrials.gov