A Osteopetrose, também conhecida como doença dos ossos de mármore, é um termo que descreve um grupo de doenças raras e hereditárias que afetam os ossos do corpo. Elas são caracterizadas por uma densidade óssea maior do que o normal, visível em exames de raio-X.
Introdução
O que você precisa saber de cara
A Osteopetrose, também conhecida como doença dos ossos de mármore, é um termo que descreve um grupo de doenças raras e hereditárias que afetam os ossos do corpo. Elas são caracterizadas por uma densidade óssea maior do que o normal, visível em exames de raio-X.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 143 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 401 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
19 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive, X-linked recessive.
Curadoria gene-doença
fontes oficiaisPlays a central role in cell adhesion in hematopoietic cells (PubMed:19234463, PubMed:26359933). Acts by activating the integrin beta-1-3 (ITGB1, ITGB2 and ITGB3) (By similarity). Required for integrin-mediated platelet adhesion and leukocyte adhesion to endothelial cells (PubMed:19234460). Required for activation of integrin beta-2 (ITGB2) in polymorphonuclear granulocytes (PMNs) (By similarity) Isoform 2 may act as a repressor of NF-kappa-B and apoptosis
Cell projection, podosome
Leukocyte adhesion deficiency 3
A disorder characterized by recurrent bacterial infections without pus formation, leukocytosis and major bleeding disorders.
Key downstream component of the canonical Wnt signaling pathway (PubMed:17524503, PubMed:18077326, PubMed:18086858, PubMed:18957423, PubMed:21262353, PubMed:22155184, PubMed:22647378, PubMed:22699938). In the absence of Wnt, forms a complex with AXIN1, AXIN2, APC, CSNK1A1 and GSK3B that promotes phosphorylation on N-terminal Ser and Thr residues and ubiquitination of CTNNB1 via BTRC and its subsequent degradation by the proteasome (PubMed:17524503, PubMed:18077326, PubMed:18086858, PubMed:189574
CytoplasmNucleusCytoplasm, cytoskeletonCell junction, adherens junctionCell junctionCell membraneCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, spindle poleSynapseCytoplasm, cytoskeleton, cilium basal body
Colorectal cancer
A complex disease characterized by malignant lesions arising from the inner wall of the large intestine (the colon) and the rectum. Genetic alterations are often associated with progression from premalignant lesion (adenoma) to invasive adenocarcinoma. Risk factors for cancer of the colon and rectum include colon polyps, long-standing ulcerative colitis, and genetic family history.
Regulatory subunit of the IKK core complex which phosphorylates inhibitors of NF-kappa-B thus leading to the dissociation of the inhibitor/NF-kappa-B complex and ultimately the degradation of the inhibitor (PubMed:14695475, PubMed:20724660, PubMed:21518757, PubMed:9751060). Its binding to scaffolding polyubiquitin plays a key role in IKK activation by multiple signaling receptor pathways (PubMed:16547522, PubMed:18287044, PubMed:19033441, PubMed:19185524, PubMed:21606507, PubMed:27777308, PubMed
CytoplasmNucleus
Ectodermal dysplasia and immunodeficiency 1
A form of ectoderma dysplasia, a heterogeneous group of disorders due to abnormal development of two or more ectodermal structures. EDAID1 is an X-linked recessive disorder characterized by absence of sweat glands, sparse scalp hair, rare conical teeth and immunological abnormalities resulting in severe infectious diseases. Severely affected individuals may also show lymphedema, osteopetrosis, and, rarely, hematologic abnormalities. The phenotype is highly variable, and may be fatal in childhood.
Tyrosine-protein kinase that acts as a cell-surface receptor for CSF1 and IL34 and plays an essential role in the regulation of survival, proliferation and differentiation of hematopoietic precursor cells, especially mononuclear phagocytes, such as macrophages and monocytes. Promotes the release of pro-inflammatory chemokines in response to IL34 and CSF1, and thereby plays an important role in innate immunity and in inflammatory processes. Plays an important role in the regulation of osteoclast
Cell membrane
Uniporter that mediates the facilitative transport of nucleoside across lysosomal and mitochondrial membranes (PubMed:15701636, PubMed:19164483, PubMed:20595384, PubMed:28729424). Functions as a non-electrogenic Na(+)-independent transporter (PubMed:15701636, PubMed:19164483, PubMed:28729424). Substrate transport is pH-dependent and enhanced under acidic condition, probably reflecting the location of the transporter in acidic intracellular compartments (PubMed:15701636, PubMed:19164483, PubMed:2
Lysosome membraneLate endosome membraneMitochondrion membraneCell membrane
Histiocytosis-lymphadenopathy plus syndrome
A syndrome characterized by the combination of features from 2 or more of four histiocytic disorders, originally thought to be distinct: Faisalabad histiocytosis (FHC), sinus histiocytosis with massive lymphadenopathy (SHML), H syndrome, and pigmented hypertrichosis with insulin-dependent diabetes mellitus syndrome (PHID). FHC features include joint deformities, sensorineural hearing loss, and subsequent development of generalized lymphadenopathy and swellings in the eyelids that contain histiocytes. SHML causes lymph node enlargement in children frequently accompanied by fever, leukocytosis, elevated erythrocyte sedimentation rate, and polyclonal hypergammaglobulinemia. H syndrome is characterized by cutaneous hyperpigmentation and hypertrichosis, hepatosplenomegaly, heart anomalies, and hypogonadism; hearing loss is found in about half of patients. PHID is characterized by predominantly antibody-negative insulin-dependent diabetes mellitus associated with pigmented hypertrichosis and variable occurrence of other features of H syndrome.
Sodium-independent anion exchanger which mediates the electroneutral exchange of chloride for bicarbonate ions across the cell membrane (PubMed:15184086, PubMed:34668226). Plays an important role in osteoclast differentiation and function (PubMed:34668226). Regulates bone resorption and calpain-dependent actin cytoskeleton organization in osteoclasts via anion exchange-dependent control of pH (By similarity). Essential for intracellular pH regulation in CD8(+) T-cells upon CD3 stimulation, modul
Apical cell membraneBasolateral cell membrane
Osteopetrosis, autosomal recessive 9
A form of osteopetrosis, a rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. OPTB9 is characterized by increased bone density and bone fragility, as well as renal failure.
Acts as a coreceptor with members of the frizzled family of seven-transmembrane spanning receptors to transduce signal by Wnt proteins (PubMed:11336703, PubMed:11448771, PubMed:11719191, PubMed:15778503, PubMed:15908424, PubMed:16252235). Activates the canonical Wnt signaling pathway that controls cell fate determination and self-renewal during embryonic development and adult tissue regeneration (PubMed:11336703, PubMed:11719191). In particular, may play an important role in the development of t
MembraneEndoplasmic reticulum
Vitreoretinopathy, exudative 1
An autosomal dominant disorder of the retinal vasculature characterized by an abrupt cessation of growth of peripheral capillaries, leading to an avascular peripheral retina. This may lead to compensatory retinal neovascularization, which is thought to be induced by hypoxia from the initial avascular insult. New vessels are prone to leakage and rupture causing exudates and bleeding, followed by scarring, retinal detachment and blindness. Clinical features can be highly variable, even within the same family. Patients with mild forms of the disease are asymptomatic, and their only disease related abnormality is an arc of avascular retina in the extreme temporal periphery. In many ways the disease resembles retinopathy of prematurity but there is no evidence of prematurity or small birth weight in the patient history.
Thiol protease involved in osteoclastic bone resorption and may participate partially in the disorder of bone remodeling. Displays potent endoprotease activity against fibrinogen at acid pH. May play an important role in extracellular matrix degradation. Involved in the release of thyroid hormone thyroxine (T4) by limited proteolysis of TG/thyroglobulin in the thyroid follicle lumen (PubMed:11082042)
LysosomeSecretedApical cell membrane
Pycnodysostosis
A rare autosomal recessive bone disorder characterized by deformity of the skull, maxilla and phalanges, osteosclerosis, and fragility of bone.
Receptor for TNFSF11/RANKL/TRANCE/OPGL; essential for RANKL-mediated osteoclastogenesis (PubMed:9878548). Its interaction with EEIG1 promotes osteoclastogenesis via facilitating the transcription of NFATC1 and activation of PLCG2 (By similarity). Involved in the regulation of interactions between T-cells and dendritic cells (By similarity)
Cell membraneMembrane raft
Familial expansile osteolysis
Rare autosomal dominant bone disorder characterized by focal areas of increased bone remodeling. The osteolytic lesions develop usually in the long bones during early adulthood. FEO is often associated with early-onset deafness and loss of dentition.
Regulator of the canonical Wnt signaling pathway. Acts by specifically binding phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2), translocating to the cell membrane and interacting with key regulators of the canonical Wnt signaling pathway, such as components of the beta-catenin destruction complex. Acts both as a positive and negative regulator of the Wnt signaling pathway, depending on the context: acts as a positive regulator by promoting LRP6 phosphorylation. Also acts as a negative regu
CytoplasmCell membraneNucleus
Osteopathia striata with cranial sclerosis
An X-linked dominant sclerosing bone dysplasia that presents in females with macrocephaly, cleft palate, facial palsy, conductive hearing loss, mild learning disabilities, sclerosis of the long bones and skull. Longitudinal striations are visible on radiographs of the long bones, pelvis, and scapulae (osteopathia striata). In males this entity is usually associated with fetal or neonatal lethality. Occasional surviving males have, in addition to hyperostosis, cardiac, intestinal, and genitourinary malformations.
Required for osteoclast and melanocyte maturation and function
Lysosome membrane
Osteopetrosis, autosomal recessive 5
A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. OPTB5 patients manifest primary central nervous system involvement in addition to the classical stigmata of severe bone sclerosis, growth failure, anemia, thrombocytopenia and visual impairment with optic atrophy.
Catalyzes the reversible hydration of carbon dioxide (PubMed:11327835, PubMed:11802772, PubMed:11831900, PubMed:12056894, PubMed:12171926, PubMed:1336460, PubMed:14736236, PubMed:15300855, PubMed:15453828, PubMed:15667203, PubMed:15865431, PubMed:16106378, PubMed:16214338, PubMed:16290146, PubMed:16686544, PubMed:16759856, PubMed:16807956, PubMed:17127057, PubMed:17251017, PubMed:17314045, PubMed:17330962, PubMed:17346964, PubMed:17540563, PubMed:17588751, PubMed:17705204, PubMed:18024029, PubMe
CytoplasmCell membrane
Osteopetrosis, autosomal recessive 3
A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. OPTB3 is associated with renal tubular acidosis, cerebral calcification (marble brain disease) and in some cases with intellectual disability.
Cytokine that binds to TNFRSF11B/OPG and to TNFRSF11A/RANK. Osteoclast differentiation and activation factor. Augments the ability of dendritic cells to stimulate naive T-cell proliferation. May be an important regulator of interactions between T-cells and dendritic cells and may play a role in the regulation of the T-cell-dependent immune response. May also play an important role in enhanced bone-resorption in humoral hypercalcemia of malignancy (PubMed:22664871). Induces osteoclastogenesis by
Cell membraneCytoplasmSecreted
Osteopetrosis, autosomal recessive 2
A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. OPTB2 is characterized by paucity of osteoclasts, suggesting a molecular defect in osteoclast development.
Acts as a multivalent adapter protein that regulates Rab7-dependent and HOPS complex-dependent fusion events in the endolysosomal system and couples autophagic and the endocytic trafficking pathways. Acts as a dual effector of RAB7A and ARL8B that simultaneously binds these GTPases, bringing about clustering and fusion of late endosomes and lysosomes (PubMed:25498145, PubMed:28325809). Required for late stages of endolysosomal maturation, facilitating both endocytosis-mediated degradation of gro
Autolysosome membraneEndosome membraneLate endosome membraneLysosome membrane
Osteopetrosis, autosomal recessive 6
A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves.
Subunit of the V0 complex of vacuolar(H+)-ATPase (V-ATPase), a multisubunit enzyme composed of a peripheral complex (V1) that hydrolyzes ATP and a membrane integral complex (V0) that translocates protons (By similarity). V-ATPase is responsible for acidifying and maintaining the pH of intracellular compartments and in some cell types, is targeted to the plasma membrane, where it is responsible for acidifying the extracellular environment (By similarity). Seems to be directly involved in T-cell a
Membrane
Osteopetrosis, autosomal recessive 1
A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves.
Slowly voltage-gated channel mediating the exchange of chloride ions against protons (PubMed:18449189, PubMed:21527911). Functions as antiporter and contributes to the acidification of the lysosome lumen and may be involved in maintaining lysosomal pH (PubMed:18449189, PubMed:21527911, PubMed:31155284). The CLC channel family contains both chloride channels and proton-coupled anion transporters that exchange chloride or another anion for protons (By similarity). The presence of conserved gating
Lysosome membrane
Osteopetrosis, autosomal recessive 4
A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves.
Serine/threonine-protein kinase which phosphorylates RAB proteins involved in intracellular trafficking (PubMed:36040231). Phosphorylates RAB7A; this activity is dependent on protein kinase C (PKC) activation (PubMed:36040231, PubMed:37558661, PubMed:37857821). Plays a role in the negative regulation of bone mass, acting through the maturation of osteoclasts (By similarity)
CytoplasmCell membrane
Osteosclerotic metaphyseal dysplasia
An autosomal recessive skeletal dysplasia characterized by osteosclerosis at multiple skeletal sites, predominantly at the metaphyses of the long bones and vertebral bodies.
Probable phosphoinositide-binding protein involved in protein sorting and membrane trafficking in endosomes. Plays a role in cilium biogenesis through regulation of the transport and the localization of proteins to the cilium. Required for the localization to the cilium of V-ATPase subunit ATP6V1D and ATP6V0D1, and RAB8A. Involved in osteoclast differentiation and therefore bone resorption
CytoplasmEndosome membraneCytoplasm, cytoskeleton, microtubule organizing center, centrosome
Osteopetrosis, autosomal recessive 8
A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. OPTB8 is clinically characterized by dense bones with no distinction between outer and inner plates, due to extensive encroachment of cortical bone into the medullary space, increased head circumference, broad open fontanelle, frontal bossing, and hepatosplenomegaly. Osteoclasts number is low and their bone resorptive capacity is impaired.
Dual specificity protein kinase which acts as an essential component of the MAP kinase signal transduction pathway. Binding of extracellular ligands such as growth factors, cytokines and hormones to their cell-surface receptors activates RAS and this initiates RAF1 activation. RAF1 then further activates the dual-specificity protein kinases MAP2K1/MEK1 and MAP2K2/MEK2. Both MAP2K1/MEK1 and MAP2K2/MEK2 function specifically in the MAPK/ERK cascade, and catalyze the concomitant phosphorylation of
Cytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, microtubule organizing center, spindle pole bodyCytoplasmNucleusMembrane
Cardiofaciocutaneous syndrome 3
A form of cardiofaciocutaneous syndrome, a multiple congenital anomaly disorder characterized by a distinctive facial appearance, heart defects and intellectual disability. Heart defects include pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Some affected individuals present with ectodermal abnormalities such as sparse, friable hair, hyperkeratotic skin lesions and a generalized ichthyosis-like condition. Typical facial features are similar to Noonan syndrome. They include high forehead with bitemporal constriction, hypoplastic supraorbital ridges, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly angulated ears with prominent helices. Distinctive features of CFC3 include macrostomia and horizontal shape of palpebral fissures.
Variantes genéticas (ClinVar)
729 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
111 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Osteopetrose
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
27 ensaios clínicos encontrados, 2 ativos.
Publicações mais relevantes
Fractures are highly correlated with bone density and inversely correlated with bone turnover markers in autosomal dominant osteopetrosis.
Autosomal dominant osteopetrosis (ADO) is a rare osteosclerotic disorder usually caused by missense variants in the CLCN7 gene, which results in impaired osteoclastic bone resorption. Penetrance is incomplete, and disease severity varies widely, even among relatives within the same family. Although ADO can cause visual loss, osteonecrosis, osteomyelitis, and bone marrow failure, the most common complication of ADO is fracture. We are conducting a natural history study to characterize disease progression and determinants of disease severity. We hypothesized that baseline BMD and bone turnover markers would correlate with self-reported fracture history. We report cross-sectional analysis of baseline data from the natural history study in 54 individuals (42 adults, 12 children). In adults, Z-scores for both volumetric (r = 0.87, p < .001) and areal BMD (aBMD) of the LS, and Z-scores for FN, and TH aBMD (r = 0.77 to 0.78; p < .001) were correlated with lifetime fracture number. Tartrate resistant acid phosphatase, a marker of osteoclast number, correlated positively with fracture (r = 0.52, p = .004) consistent with an adaptive response of higher numbers of osteoclasts among more severely affected individuals. However, fracture number correlated inversely with the bone resorption markers serum C-telopeptide (r = -0.60, p < .001) and urine N-telopeptide/creatinine ratio (r = -0.35, p = .047), suggesting that ADO subjects who have the most reduced osteoclast activity have a greater tendency to fracture. Correlation coefficients between fractures, BMD, and bone turnover markers were similar when limited to the 37 adults with disease-causing CLCN7 variants. There were no statistically significant differences between subjects with the most common CLCN7 variant (G215R), the most common variant in our cohort, compared to other CLCN7 variants with respect to fracture, bone density measures, or biochemical markers of bone turnover. These data demonstrate that bone density and biochemical bone turnover markers are indicators of ADO severity as defined by fracture number. Autosomal dominant osteopetrosis (ADO) is a rare genetic bone disorder characterized by abnormally dense but fragile bones. The severity of the condition can vary significantly, even among individuals within the same family. This natural history study aims to better understand the progression of ADO and identify factors that influence disease severity. Unlike osteoporosis, individuals with ADO who exhibit the highest bone density are paradoxically at the greatest risk for fractures. Furthermore, lower levels of biochemical markers of bone turnover were associated with both increased bone density and a higher incidence of fractures. Importantly, the severity of ADO did not correlate with the specific genetic variants responsible for the condition. In summary, ADO patients with the highest bone density have the most fractures. These findings suggest that both bone density and biochemical markers of bone turnover can serve as meaningful clinical endpoints in future trials evaluating potential therapies for ADO. TCIRG1-related osteopetrosis is characterized by growth deficiency, pathologic fractures of dense but brittle bones, limping gait with bone pain, hypocalcemia that can result in seizures, and secondary hyperparathyroidism. Advanced bone sclerosis results in extramedullary hematopoiesis, bone marrow failure, ocular complications with potential for blindness (optic nerve compression/atrophy and primary retinopathy), dental manifestations (delay in tooth eruption and dental caries), and deafness in some individuals. Craniofacial features can include macrocephaly, exophthalmos, hypertelorism, and micrognathia. There is wide variability in clinical severity. Severe osteopetrosis with infantile onset is the most common phenotype, potentially resulting in death at a young age in the absence of successful treatment. Individuals with mild TCIRG1-related osteopetrosis may have normal growth without hematologic or neurologic abnormalities. The diagnosis of TCIRG1-related osteopetrosis is established in a proband with characteristic clinical, laboratory, and imaging findings and biallelic pathogenic variants in TCIRG1 identified by molecular genetic testing. Note: Heterozygous TCIRG1 pathogenic variants have been rarely reported in individuals with TCIRG1-related osteopetrosis. Targeted therapies: Hematopoietic stem cell therapy (HSCT) for those with hematologic failure with imminent vision loss, severe osteopetrosis with bone marrow failure, or severe osteopetrosis in children younger than age one year, ideally prior to age ten months. Interferon gamma-1b (IFN-γ1b) may be considered in infantile TCIRG1-related osteopetrosis, to serve as a bridge to HSCT. Supportive care: Multidisciplinary management of complications includes treatment of fractures, skeletal deformities, and delayed healing per orthopedist; treatment of respiratory compromise per pulmonologist; calcium and vitamin D supplementation per endocrinologist; treatment of bone marrow failure and red blood cell transfusions per hematologist; dental treatment; management of hydrocephalus per neurosurgeon; treatment of hypocalcemia to prevent seizures; treatment of non-hypocalcemic seizures per neurologist; developmental and educational support; feeding therapy may become essential, requiring insertion of gastrostomy tube; optic nerve decompression and optic nerve sheath fenestration per neuro-ophthalmologist; surgical intervention for deafness per otolaryngologist; treatment of chronic/recurrent infections per infectious disease specialist; family and social work support. Surveillance: Assess for frequency of fractures, skeletal deformities, and bone pain at each visit; assess for respiratory compromise and frequency of infections in those with small-volume thorax; serum calcium and phosphate every six to 12 months and every three months in those on calcitriol therapy; 25-hydroxyvitamin D and intact parathyroid hormone every six to 12 months; serum creatinine, calculation of glomerular filtration rate, and urinary calcium-to-creatinine ratio every three months in those on calcitriol therapy; renal ultrasound annually in those on calcitriol therapy; complete blood count with differential every six to 12 months; dental evaluation more frequently than every six months; neurologic exam with EEG if seizures are suspected every six months; head imaging per neurologist; assess developmental progress, educational needs, feeding, and growth at each visit; ophthalmology evaluation every six months until age 18 years, then annually through adulthood, with visual field exams in those who are old enough to participate and MRI of the optic nerves as clinically indicated; annual audiology evaluation in childhood and as needed in adults; assess family and social work needs at each visit. Evaluation of relatives at risk: It is appropriate to clarify the genetic status of apparently asymptomatic sibs of a proband with TCIRG1-related osteopetrosis caused by missense variants; sibs found to be heterozygous for a TCIRG1 missense variant should be monitored for manifestations of osteopetrosis. TCIRG1-related osteopetrosis is typically inherited in an autosomal recessive manner. Note: Heterozygous TCIRG1 pathogenic variants have been identified in affected individuals in one family with autosomal dominant osteopetrosis and one additional individual with a heterozygous de novo pathogenic variant. If both parents are known to be heterozygous for a TCIRG1 pathogenic variant, each sib of an affected individual has at conception a 25% chance of having TCIRG1-related osteopetrosis, a 50% chance of being heterozygous, and a 25% chance of inheriting neither of the familial TCIRG1 pathogenic variants. To date, manifestations of TCIRG1-related osteopetrosis have not been reported in heterozygous family members of a proband with TCIRG1-related osteopetrosis; however, it is theoretically possible that heterozygous sibs may be at risk for TCIRG1-related osteopetrosis. Once the TCIRG1 pathogenic variants have been identified in an affected family member, heterozygote testing for at-risk family members and prenatal/preimplantation genetic testing are possible.
Tcirg1 deficiency delays osteoarthritis progression by impairing lysosome acidification and peripheral accumulation in osteoclasts.
Osteoarthritis (OA) is a chronic degenerative joint disease characterized by articular cartilage loss and aberrant subchondral bone remodeling. "T-cell immune regulator 1" (Tcirg1), which encodes the a3 subunit of the V-ATPase, has been demonstrated to inhibit the formation of large osteoclasts by reducing intracellular calcium oscillations. Mutations in the Tcirg1 gene sequence have been associated with osteopetrosis by impairing lysosomal transport in osteoclasts. This study aims to assess the impact of Tcirg1 on OA progression and to explore its therapeutic potential for the disease treatment. Proteomic comparison of weight-bearing region (WBR) versus non-weight-bearing region (NWBR) of the subchondral bone was performed in 20 OA patients undergoing total knee arthroplasty. OA was then surgically induced in wild-type and Tcirg1-knockout mice by destabilization of the medial meniscus; disease severity and subchondral bone architecture were evaluated by histology and micro-CT. In vitro, primary bone marrow macrophages were differentiated into osteoclasts to assess the role of Tcirg1 in osteoclastogenesis, focusing on cell fusion, bone resorption, and lysosome acidification and distribution. Proteomic analysis revealed that TCIRG1 was significantly upregulated in the WBR compared to NWBR of subchondral bone in OA patients, with functional enrichment analysis indicating TCIRG1 correlation with lysosome-related biological processes. In the murine OA model, Tcirg1 expression increased in parallel with osteoclast activity, peaking at 4 weeks post-surgery, which coincided with severe subchondral bone loss. Tcirg1 deficiency in knockout mice delayed OA progression, as evidenced by reduced cartilage damage, improved subchondral bone mass, and decreased osteoclast activity. In vitro, Tcirg1 expression increased during osteoclast differentiation, and its knockdown inhibited osteoclast fusion and bone resorption by impairing lysosome acidification and peripheral accumulation. Tcirg1 regulates lysosome acidification and peripheral accumulation, thereby influencing osteoclast activity in the subchondral bone. Given that Tcirg1 knockdown was found to slow down the progression of OA, targeting Tcirg1 may serve as a potential therapeutic strategy for treating OA.
In vivo haemopoietic stem cell gene therapy enabled by postnatal trafficking.
Lentiviral vector (LV)-mediated ex vivo gene therapy for haematopoietic stem and progenitor cells (HSPCs) has delivered on the promise of a 'one-and-done' treatment for several genetic diseases1. However, ex vivo manipulation and patient conditioning before transplantation are major hurdles that could be overcome by an in vivo approach. Here we demonstrate that in vivo gene delivery to HSPCs after systemic LV administration is enabled by the substantial trafficking of these cells from the liver to the bone marrow in newborn mice. We improved gene-transfer efficiency using a phagocytosis-shielded LV, successfully reaching bona fide HSPCs capable of long-term multilineage output and engraftment after serial transplantation, as confirmed by clonal tracking. HSPC mobilization further increased gene transfer, extending the window of intervention, although permissiveness to LV transduction declined with age. We successfully tested this in vivo strategy in mouse models of adenosine deaminase deficiency, autosomal recessive osteopetrosis and Fanconi anaemia. Interestingly, in vivo gene transfer provided a selective advantage to corrected HSPCs in Fanconi anaemia, leading to near-complete haematopoietic reconstitution and prevention of bone marrow failure. Given that circulating HSPCs in humans are also most abundant shortly after birth, in vivo HSPC gene transfer holds strong translational potential across multiple diseases.
Identification of a novel mutation in the CLCN7 gene in pediatric osteopetrosis: case report.
Osteopetrosis, also known as osteosclerosis and marble-bone disease, is a rare genetic metabolic bone disorder caused by the dysplasia or dysfunction of osteoclasts, usually caused by variants of chloride voltage-gated channel 7 (CLCN7) gene. We retrospectively analyzed the clinical data of two children with osteopetrosis and their families. Whole-exome sequencing (WES) was used for genetic analysis, and Sanger sequencing confirmed possible pathogenic variants. In family 1, the proband harbored a novel mutation c.2351G>C (p.R784T) in CLCN7 gene. The initial symptom of proband 1 was a post-traumatic fracture, and imaging features was "sandwich cake" -like changes. In family 2, the proband harbored previously reported compound heterozygous variants in CLCN7 gene: c.899C>T (p.A300V) and c.1534G>A (p.G512R). Among them, c.1534G>A (p.G512R) was only recorded in clinvar and no reports of protein function prediction. The initial symptom of proband 2 was cough, and imaging features was "sandwich vertebrae". Our study expands the mutation spectrum of the CLCN7 gene and provides new insights into the pathogenesis of osteopetrosis.
The correlation of intracranial parenchymal calcium score and the severity of neurological clinical presentation in carbonic anhydrase deficiency type 2.
Carbonic anhydrase type II deficiency (CAII-D) syndrome is a rare autosomal recessive genetic disorder characterized by osteopetrosis, renal tubular acidosis, and brain calcifications. Understanding the clinical and radiological features of CAII-D is key to effective management. This study aimed to comprehensively analyze and measure intracranial parenchymal calcium score in pediatric CAII-D in relation to the severity of neurological clinical presentation. A retrospective chart review at King Saud University Medical City included pediatric CAII-D patients diagnosed between June 2015 and December 2022. Study variables included age, gender, genetic results, developmental status, developmental quotient (DQ), CT findings, optic canal diameter, intracranial calcium score, and neuropsychiatric symptoms. Ten CAII-D patients, median age 10.5 years, were included. Most patients displayed homozygous pathogenic CA2 gene variants. For neurodevelopmental symptoms, 60.0 % of patients presented with global developmental delay, 20.0 % had intellectual disability, and the remaining 20.0 % had normal development. The median DQ score was 63.50, with 80.0 % categorized as delayed. Neuropsychiatric disorders were present in 20.0 %. Optic nerve atrophy was observed in 22.2 %, while brain calcifications were present in 70.0 % of cases. Correlation analysis revealed no significant associations between intracranial parenchymal calcium score and age, DQ score, or optic canal diameter. Neurodevelopmental symptoms, neuropsychiatric symptoms, and DQ were not associated with intracranial parenchymal calcium score. Intraparenchymal calcifications in CAII-D are common but unrelated to developmental delay and neuropsychiatric symptoms, suggesting complex pathophysiology. Follow-up brain imaging may not aid in prognosis or severity assessment, highlighting the need for further research.
Publicações recentes
Ver todas no PubMed📚 EuropePMC63 artigos no totalmostrando 169
Osteopetrosis misdiagnosed as congenital cytomegalovirus infection: A case report and literature review.
MedicineTcirg1 deficiency delays osteoarthritis progression by impairing lysosome acidification and peripheral accumulation in osteoclasts.
Frontiers in cell and developmental biologyFractures are highly correlated with bone density and inversely correlated with bone turnover markers in autosomal dominant osteopetrosis.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchUnderstanding rickets in osteopetrosis via a case: mechanisms and treatment implications.
Journal of pediatric endocrinology & metabolism : JPEMNeuroaxonal Dystrophy With Osteopetrosis Associated With a Novel Biallelic Nonsense Homozygous Variant in BORCS5.
American journal of medical genetics. Part ARenal tubular acidosis: Varied aetiologies and clinical presentations: Three case reports.
World journal of nephrologyRadiological diagnosis of infantile osteopetrosis in a 1-year-old with macrocephaly and jaundice.
Radiology case reportsIn vivo haemopoietic stem cell gene therapy enabled by postnatal trafficking.
NatureBilateral Endoscopic Endonasal Optic Nerve Decompression in an Infant with Osteopetrosis: A Case Report.
Journal of neurological surgery reportsIdentification of a novel mutation in the CLCN7 gene in pediatric osteopetrosis: case report.
Frontiers in pediatricsA novel frameshift variant leads to familial osteopetrosis with variable phenotypes in a Chinese Han consanguineous family.
BMC medical genomicsEvaluation of Quality and Bone Microarchitecture Alterations in Osteopetrosis Patients: Assessed by HR-PQCT.
The Journal of clinical endocrinology and metabolismTotal shoulder arthroplasty in a patient with osteopetrosis: A case report.
Radiology case reportsThe correlation of intracranial parenchymal calcium score and the severity of neurological clinical presentation in carbonic anhydrase deficiency type 2.
Brain & developmentTherapeutic targeting of Wnt antagonists by small molecules for treatment of osteoporosis.
Biochemical pharmacologyMalignant osteopetrosis of infancy: A case report.
International journal of surgery case reportsChallenges of Hip and Knee Arthroplasty in Patients With Osteopetrosis.
The Journal of the American Academy of Orthopaedic SurgeonsMolecular Heterogeneity of Osteopetrosis in India: Report of 17 Novel Variants.
Indian journal of hematology & blood transfusion : an official journal of Indian Society of Hematology and Blood TransfusionImpdh2 deficiency suppresses osteoclastogenesis through mitochondrial oxidative phosphorylation and alleviates ovariectomy-induced osteoporosis.
Biochemical and biophysical research communicationsGain-of-function variants in CLCN7 cause hypopigmentation and lysosomal storage disease.
The Journal of biological chemistryCharacterization of a spontaneous osteopetrosis model using RANKL-dysfunctional mice.
Tissue & cellPatient-Reported Outcomes in Autosomal Dominant Osteopetrosis: Findings From the Osteopetrosis Registry Study.
The Journal of clinical endocrinology and metabolismAllogenic hematopoietic stem cell transplantation in an Iranian patient with osteopetrosis caused by carbonic anhydrase II deficiency: A case report.
Pediatric transplantationIdentification of an novel genetic variant associated with osteoporosis: insights from the Taiwan Biobank Study.
JBMR plusOsteopetrosis and related osteoclast disorders in adults: A review and knowledge gaps On behalf of the European calcified tissue society and ERN BOND.
European journal of medical geneticsFosl2 Deficiency Predisposes Mice to Osteopetrosis, Leading to Bone Marrow Failure.
Journal of immunology (Baltimore, Md. : 1950)Osteopetrosis complicated by multilevel spondylolysis.
Radiology case reportsAutosomal Dominant Osteopetrosis (ADO) Caused by a Missense Variant in the TCIRG1 Gene.
The Journal of clinical endocrinology and metabolismSuccessful complete oral rehabilitation of a patient with osteopetrosis with extensive pre-treatments, bone grafts, dental implants and fixed bridges: a multidisciplinary case report.
BMC oral healthCranial distraction osteogenesis for craniosynostosis associated with osteopetrosis: A case report.
Surgical neurology internationalDistinct ClC-6 and ClC-7 Cl- sensitivities provide insight into ClC-7's role in lysosomal Cl- homeostasis.
The Journal of physiologyFluconazole-Induced Protein Changes in Osteogenic and Immune Metabolic Pathways of Dental Pulp Mesenchymal Stem Cells of Osteopetrosis Patients.
International journal of molecular sciencesThe molecular spectrum of Turkish osteopetrosis and related osteoclast disorders with natural history, including a candidate gene, CCDC120.
BoneNovel hybrid silicon-lipid nanoparticles deliver a siRNA to cure autosomal dominant osteopetrosis in mice. Implications for gene therapy in humans.
Molecular therapy. Nucleic acidsLRP5 high bone mass (Worth-type autosomal dominant endosteal hyperostosis): case report and historical review of the literature.
Archives of osteoporosisOsteosclerotic Metaphyseal Dysplasia Due to a Likely Pathogenic LRRK1 Variant as a Cause of Recurrent Long Bone Fractures.
JBMR plusCRISPR/Cas9-Mediated Gene Correction in Osteopetrosis Patient-Derived iPSCs.
Frontiers in bioscience (Landmark edition)Molecular Mechanisms of Craniofacial and Dental Abnormalities in Osteopetrosis.
International journal of molecular sciencesBrain abnormalities, neurodegeneration, and dysosteosclerosis (BANDDOS): new cases, systematic literature review, and associations with CSF1R-ALSP.
Orphanet journal of rare diseasesGNAS gene mutations affecting XLαs and bone health: A long neglected relationship.
Clinical geneticsLRP6 High Bone Mass Characterized in Two Generations Harboring a Unique Mutation of Low-Density Lipoprotein Receptor-Related Protein 6.
JBMR plusClinical and Osteopetrosis-Like Radiological Findings in Patients with Leukocyte Adhesion Deficiency Type III.
Journal of clinical immunologyCase report: Gene mutations and clinical characteristics of four patients with osteopetrosis.
Frontiers in pediatricsAutosomal dominant osteopetrosis.
BoneClinical and genetic diagnosis of autosomal dominant osteopetrosis type II in a Chinese family: A case report.
World journal of clinical casesOne half-century of advances in the evaluation and management of disorders of bone and mineral metabolism in children and adolescents.
Journal of pediatric endocrinology & metabolism : JPEMCLCN7, a gene shared by autosomal recessive and autosomal dominant osteopetrosis.
BoneOsteopetrosis: The patient point of view and medical challenges.
BoneSpectrum of Skeletal Imaging Features in Osteopetrosis: Inheritance Pattern and Radiological Associations.
GenesImaging in osteopetrosis.
BoneBone marrow transplantation as a therapy for autosomal dominant osteopetrosis type 2 in mice.
FASEB journal : official publication of the Federation of American Societies for Experimental BiologyCase report of mild TCIRG1-associated autosomal recessive osteopetrosis in Vietnam.
American journal of medical genetics. Part AAutoimmune Cytopenias Post Hematopoietic Stem Cell Transplantation in Pediatric Patients With Osteopetrosis and Other Nonmalignant Diseases.
Frontiers in immunologyTyrosine Kinase Src Is a Regulatory Factor of Bone Homeostasis.
International journal of molecular sciencesNatural History of Type II Autosomal Dominant Osteopetrosis: A Single Center Retrospective Study.
Frontiers in endocrinologyOpen-Label Pilot Study of Interferon Gamma-1b in Patients With Non-Infantile Osteopetrosis.
JBMR plusThe Treatment of Subtrochanteric Fracture with Reversed Contralateral Distal Femoral Locking Compression Plate (DF-LCP) Using a Progressive and Intermittent Drilling Procedure in Three Osteopetrosis Patients.
Orthopaedic surgeryMolecular structure, expression, and the emerging role of Siglec-15 in skeletal biology and cancer.
Journal of cellular physiologyCircular RNAs as potential regulators in bone remodeling: a narrative review.
Annals of translational medicineBroadening the phenotype of LRRK1 mutations - Features of malignant osteopetrosis and optic nerve atrophy with intrafamilial variable expressivity.
European journal of medical geneticsOverlapping Phenotypes in Osteopetrosis and Pycnodysostosis in Asian-Indians.
Case reports in geneticsSLC4A2 Deficiency Causes a New Type of Osteopetrosis.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchNew Insights Into Osteoclast Biology.
JBMR plusA successful implant-supported fixed prosthesis in a patient with osteopetrosis: A clinical report.
The Journal of prosthetic dentistryA cross-sectional nationwide survey of osteosclerotic skeletal dysplasias in Japan.
Journal of orthopaedic science : official journal of the Japanese Orthopaedic AssociationHaploidentical haematopoietic stem cell transplantation for malignant infantile osteopetrosis and intermediate osteopetrosis: a retrospective analysis of a single centre.
Orphanet journal of rare diseasesA novel mutation in TNFRSF11A gene causes pediatric osteopetrosis: case report.
BMC surgeryManaging challenging pain and irritability in OSTM1 mutation-related infantile malignant osteopetrosis.
BMJ case reportsAutosomal recessive osteopetrosis: mechanisms and treatments.
Disease models & mechanismsEfficient generation of osteoclasts from human induced pluripotent stem cells and functional investigations of lethal CLCN7-related osteopetrosis.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchFurther expanding the mutational spectrum of brain abnormalities, neurodegeneration, and dysosteosclerosis: A rare disorder with neurologic regression and skeletal features.
American journal of medical genetics. Part AInsertion Mutation in Tnfrsf11a Causes a Paget's Disease-Like Phenotype in Heterozygous Mice and Osteopetrosis in Homozygous Mice.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchNeurodegeneration Upon Dysfunction of Endosomal/Lysosomal CLC Chloride Transporters.
Frontiers in cell and developmental biologyFurther understanding on osteopetrotic femoral fractures: a case report and literature review.
BMC surgeryThe French multicentre elevated bone mass study: prevalence and causes.
Osteoporosis international : a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USAHematopoietic stem cell transplantation in a patient with osteopetrosis and mutation in CLCN7: long-term follow-up.
Boletin medico del Hospital Infantil de MexicoCCDC154 Mutant Caused Abnormal Remodeling of the Otic Capsule and Hearing Loss in Mice.
Frontiers in cell and developmental biologyWest Syndrome Caused By a Chloride/Proton Exchange-Uncoupling CLCN6 Mutation Related to Autophagic-Lysosomal Dysfunction.
Molecular neurobiologyGene therapy for infantile malignant osteopetrosis: review of pre-clinical research and proof-of-concept for phenotypic reversal.
Molecular therapy. Methods & clinical developmentSkeletal Biology and Disease Modeling in Zebrafish.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchProteomic Profiling of the First Human Dental Pulp Mesenchymal Stem/Stromal Cells from Carbonic Anhydrase II Deficiency Osteopetrosis Patients.
International journal of molecular sciencesThe molecular structure and function of sorting nexin 10 in skeletal disorders, cancers, and other pathological conditions.
Journal of cellular physiologyLarge transient capacitive currents in wild-type lysosomal Cl-/H+ antiporter ClC-7 and residual transport activity in the proton glutamate mutant E312A.
The Journal of general physiologyPathobiologic Mechanisms of Neurodegeneration in Osteopetrosis Derived From Structural and Functional Analysis of 14 ClC-7 Mutants.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchHaploidentical transplantation in pediatric non-malignant diseases: A retrospective analysis on behalf of the Spanish Group for Hematopoietic Transplantation (GETH).
European journal of haematologyA missense mutation sheds light on a novel structure-function relationship of RANKL.
Journal of cellular physiologyMolecular insights into the human CLC-7/Ostm1 transporter.
Science advancesHigh bone mass from mutation of low-density lipoprotein receptor-related protein 6 (LRP6).
BoneSuccessful total hip arthroplasty for autosomal dominant osteopetrosis complicated by hip osteoarthritis: A case report and review of the literature.
Experimental and therapeutic medicineAdult Osteosclerotic Metaphyseal Dysplasia With Progressive Osteonecrosis of the Jaws and Abnormal Bone Resorption Pattern Due to a LRRK1 Splice Site Mutation.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchA Homozygous Mutation in 5' Untranslated Region of TNFRSF11A Leading to Molecular Diagnosis of Osteopetrosis Coinheritance With Wiskott-Aldrich Syndrome.
Journal of pediatric hematology/oncologyExpanded circulating hematopoietic stem/progenitor cells as novel cell source for the treatment of TCIRG1 osteopetrosis.
HaematologicaKif1c regulates osteoclastic bone resorption as a downstream molecule of p130Cas.
Cell biochemistry and functionHematopoietic stem cell transplantation-induced bone remodeling in autosomal recessive osteopetrosis: Interaction between skeleton and hematopoietic and sensory nervous systems.
BoneA novel homozygous LRRK1 stop gain mutation in a patient suspected with osteosclerotic metaphyseal dysplasia.
Annals of human geneticsOsteoclastogenesis inhibition by mutated IGSF23 results in human osteopetrosis.
Cell proliferationLessons Learned from Long-Term Management of Hip Fracture in Patients with Osteopetrosis: A Report of Nine Hips in Five Patients.
Journal of bone metabolismGenotyping, generation and proteomic profiling of the first human autosomal dominant osteopetrosis type II-specific induced pluripotent stem cells.
Stem cell research & therapyOsteopetrotic induced pluripotent stem cells derived from patients with different disease-associated mutations by non-integrating reprogramming methods.
Stem cell research & therapyImmune Function and Diversity of Osteoclasts in Normal and Pathological Conditions.
Frontiers in immunologyChitosan/poly(γ-glutamic acid) nanoparticles incorporating IFN-γ for immune response modulation in the context of colorectal cancer.
Biomaterials scienceExtra-skeletal manifestations in mice affected by Clcn7-dependent autosomal dominant osteopetrosis type 2 clinical and therapeutic implications.
Bone researchNovel c.G630A TCIRG1 mutation causes aberrant splicing resulting in an unusually mild form of autosomal recessive osteopetrosis.
Journal of cellular biochemistryNew explanation for autosomal dominant high bone mass: Mutation of low-density lipoprotein receptor-related protein 6.
BoneTCIRG1 and SNX10 gene mutations in the patients with autosomal recessive osteopetrosis.
GeneStem cell transplantation for osteopetrosis in patients beyond the age of 5 years.
Blood advancesHaploidentical Hematopoietic Stem Cell Transplantation with Post-Transplant Cyclophosphamide for Primary Immunodeficiencies and Inherited Disorders in Children.
Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow TransplantationClC-7 Regulates the Pattern and Early Development of Craniofacial Bone and Tooth.
TheranosticsGeneration of a human induced pluripotent stem cell line (BIHi002-A) from a patient with CLCN7-related infantile malignant autosomal recessive osteopetrosis.
Stem cell researchRole of Semaphorins in Immunopathologies and Rheumatic Diseases.
International journal of molecular sciencesCrocin attenuates methylglyoxal-induced osteoclast dysfunction by regulating glyoxalase, oxidative stress, and mitochondrial function.
Food and chemical toxicology : an international journal published for the British Industrial Biological Research AssociationPotential oligogenic disease of mental retardation, short stature, spastic paraparesis, and osteopetrosis.
The application of clinical geneticsSclerosing bone dysplasias.
Best practice & research. Clinical endocrinology & metabolismCLCN7 and TCIRG1 mutations in a single family: Evidence for digenic inheritance of osteopetrosis.
Molecular medicine reportsGenetic Risk Factors for Atypical Femoral Fractures (AFFs): A Systematic Review.
JBMR plusGenetic Analysis of CLCN7 in an Old Female Patient with Type II Autosomal Dominant Osteopetrosis.
Endocrinology and metabolism (Seoul, Korea)LncRNA AK077216 promotes RANKL-induced osteoclastogenesis and bone resorption via NFATc1 by inhibition of NIP45.
Journal of cellular physiologySclerosteosis: Report of type 1 or 2 in three Indian Tamil families and literature review.
BoneMiT family translocation renal cell carcinoma after malignant infantile osteopetrosis in childhood: a case report.
International journal of clinical and experimental pathologyWhole exome sequencing identified two novel homozygous missense variants in the same codon of CLCN7 underlying autosomal recessive infantile malignant osteopetrosis in a Pakistani family.
Molecular biology reportsCLC Chloride Channels and Transporters: Structure, Function, Physiology, and Disease.
Physiological reviewsCase Report of Worth Syndrome and Chiari I Malformation: Unusual Association and Surgical Treatment.
World neurosurgerySuccessful hematopoietic stem cell transplantation for osteopetrosis using reduced intensity conditioning.
Pediatric blood & cancerHigh bone mass in adults.
Joint bone spineMouse models of SLC4-linked disorders of HCO3--transporter dysfunction.
American journal of physiology. Cell physiologyKey Triggers of Osteoclast-Related Diseases and Available Strategies for Targeted Therapies: A Review.
Frontiers in medicineBone quality changes associated with aging and disease: a review.
Annals of the New York Academy of SciencesIdentification and in silico characterization of a novel p.P208PfsX1 mutation in V-ATPase a3 subunit associated with autosomal recessive osteopetrosis in a Pakistani family.
BMC medical geneticsA ERK/RSK-mediated negative feedback loop regulates M-CSF-evoked PI3K/AKT activation in macrophages.
FASEB journal : official publication of the Federation of American Societies for Experimental BiologyClinical Significance of DXA and HR-pQCT in Autosomal Dominant Osteopetrosis (ADO II).
Calcified tissue internationalIFN-γ alters the expression of diverse immunity related genes in a cell culture model designed to represent maturing neutrophils.
PloS oneDecompressive Cranioplasty in a Patient with Osteopetrosis.
World neurosurgeryNovel mutations of TCIRG1 cause a malignant and mild phenotype of autosomal recessive osteopetrosis (ARO) in four Chinese families.
Acta pharmacologica Sinica[Analysis of TCIRG1 gene mutation in a Chinese family affected with infantile malignant osteopetrosis].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsGuided growth for valgus deformity correction of knees in a girl with osteopetrosis: a case report.
Strategies in trauma and limb reconstructionSerum CTX levels and histomorphometric analysis in Src versus RANKL knockout mice.
Journal of bone and mineral metabolismPeriodontal profile and radiographic characterization of the jaws in a patient with autosomal dominant osteopetrosis.
Endocrinology, diabetes & metabolism case reportsClC Channels and Transporters: Structure, Physiological Functions, and Implications in Human Chloride Channelopathies.
Frontiers in pharmacologyAutozygosity reveals recessive mutations and novel mechanisms in dominant genes: implications in variant interpretation.
Genetics in medicine : official journal of the American College of Medical GeneticsRole and mechanism of action of leucine-rich repeat kinase 1 in bone.
Bone researchCase Report of Clinical Vignette: Osteopetrosis.
Military medicineHematopoietic stem cell transplantation corrects osteopetrosis in a child carrying a novel homozygous mutation in the FERMT3 gene.
BoneThe Formation of Calcified Nanospherites during Micropetrosis Represents a Unique Mineralization Mechanism in Aged Human Bone.
Small (Weinheim an der Bergstrasse, Germany)Raine Syndrome (OMIM #259775), Caused By FAM20C Mutation, Is Congenital Sclerosing Osteomalacia With Cerebral Calcification (OMIM 259660).
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchHomozygous deletion of RAG1, RAG2 and 5' region TRAF6 causes severe immune suppression and atypical osteopetrosis.
Clinical geneticsPLEKHM1/DEF8/RAB7 complex regulates lysosome positioning and bone homeostasis.
JCI insightLysosomal Ca2+ Signaling is Essential for Osteoclastogenesis and Bone Remodeling.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchAutosomal dominant osteopetrosis associated with renal tubular acidosis is due to a CLCN7 mutation.
American journal of medical genetics. Part AGenetics of osteoporosis: searching for candidate genes for bone fragility.
Archives of endocrinology and metabolismNovel CLCN7 mutation identified in a Han Chinese family with autosomal dominant osteopetrosis-2.
Molecular painCharacterization of a Relatively Malignant Form of Osteopetrosis Caused by a Novel Mutation in the PLEKHM1 Gene.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchThe use of whole exome sequencing for the diagnosis of autosomal recessive malignant infantile osteopetrosis.
Clinical geneticsOral Rehabilitation of an Osteopetrosis Patient with Osteomyelitis.
Case reports in dentistryIdiopathic Acquired Osteosclerosis in a Middle-Aged Woman With Systemic Lupus Erythematosus.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchOsteopetrosis in two siblings: two case reports.
BMC research notesLoss of PPARγ in endothelial cells leads to impaired angiogenesis.
Journal of cell scienceImproved Outcomes of Hematopoietic Stem Cell Transplantation in Patients With Infantile Malignant Osteopetrosis Using Fludarabine-Based Conditioning.
Pediatric blood & cancerA novel mutation and a known mutation in the CLCN7 gene associated with relatively stable infantile malignant osteopetrosis in a Chinese patient.
GenePotential blindness in children of patients with hereditary bone disease.
Osteoporosis international : a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USADeficiency of sorting nexin 10 prevents bone erosion in collagen-induced mouse arthritis through promoting NFATc1 degradation.
Annals of the rheumatic diseasesTwo novel mutations of CLCN7 gene in Chinese families with autosomal dominant osteopetrosis (type II).
Journal of bone and mineral metabolismA tale of two CLCs: biophysical insights toward understanding ClC-5 and ClC-7 function in endosomes and lysosomes.
The Journal of physiologyHematopoietic stem cell transplantation for infantile osteopetrosis.
BloodThe clinical spectrum and pathophysiology of skeletal complications in lysosomal storage disorders.
Best practice & research. Clinical endocrinology & metabolismNovel targets for the prevention of osteoporosis - lessons learned from studies of metabolic bone disorders.
Expert opinion on therapeutic targetsNon-total body irradiation myeloablative conditioning with intravenous busulfan and cyclophosphamide in hematopoietic stem cell transplantation for malignant infantile osteopetrosis.
Pediatric transplantationAn Adolescent Case of Osteopetrosis with Portal Hypertension as well as Mandibula Osteomyelitis.
Indian journal of hematology & blood transfusion : an official journal of Indian Society of Hematology and Blood TransfusionTwo novel CAII mutations causing carbonic anhydrase II deficiency syndrome in two unrelated Chinese families.
Metabolic brain diseaseSHP2 regulates osteoclastogenesis by promoting preosteoclast fusion.
FASEB journal : official publication of the Federation of American Societies for Experimental BiologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Osteopetrose.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Osteopetrose
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Fractures are highly correlated with bone density and inversely correlated with bone turnover markers in autosomal dominant osteopetrosis.Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research· 2026· PMID 40913471mais citado
- Tcirg1 deficiency delays osteoarthritis progression by impairing lysosome acidification and peripheral accumulation in osteoclasts.
- In vivo haemopoietic stem cell gene therapy enabled by postnatal trafficking.
- Identification of a novel mutation in the CLCN7 gene in pediatric osteopetrosis: case report.
- The correlation of intracranial parenchymal calcium score and the severity of neurological clinical presentation in carbonic anhydrase deficiency type 2.
- [Function of the CLC chloride channels and their implication in human pathology].
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2781(Orphanet)
- MONDO:0017198(MONDO)
- GARD:4155(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q1755568(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
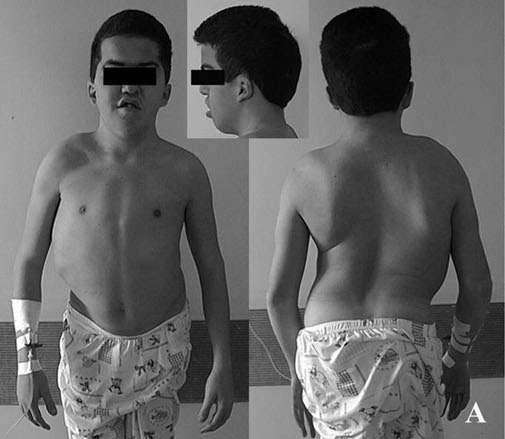
Osteopetrose
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov