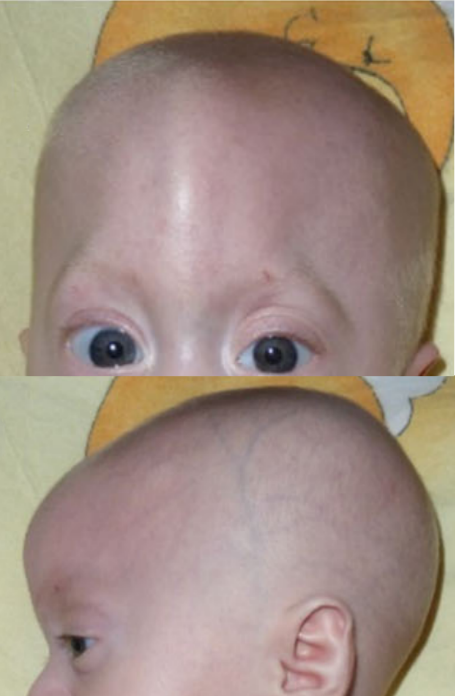
A Síndrome de C é uma condição rara que combina diversas alterações físicas presentes desde o nascimento e deficiência intelectual. As características incluem: um formato triangular da cabeça (causado pelo fechamento precoce de uma das 'emendas' ósseas na testa, chamada sutura metópica); traços faciais incomuns; pescoço curto; alterações nos ossos; e deficiência intelectual, que pode variar de leve a grave.
Introdução
O que você precisa saber de cara
A Síndrome de C é uma condição rara que combina diversas alterações físicas presentes desde o nascimento e deficiência intelectual. As características incluem: um formato triangular da cabeça (causado pelo fechamento precoce de uma das 'emendas' ósseas na testa, chamada sutura metópica); traços faciais incomuns; pescoço curto; alterações nos ossos; e deficiência intelectual, que pode variar de leve a grave.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 23 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 79 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Not applicable, Unknown.
May be involved in adhesive interactions of activated T and NK cells during the late phase of the immune response. Promotes NK cell-target adhesion by interacting with PVR present on target cells. May function at a time after T and NK cells have penetrated the endothelium using integrins and selectins, when they are actively engaging diseased cells and moving within areas of inflammation
Membrane
C syndrome
A syndrome characterized by trigonocephaly, severe intellectual disability, hypotonia, variable cardiac defects, redundant skin, and dysmorphic facial features, including upslanted palpebral fissures, epicanthal folds, depressed nasal bridge, and low-set, posteriorly rotated ears.
Variantes genéticas (ClinVar)
28 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 19 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome C
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
Pesquisa e ensaios clínicos
5 ensaios clínicos encontrados.
Publicações mais relevantes
Unveiling Mucopolysaccharidosis IIIC in Brazil: Diagnostic Journey and Clinical Features of Brazilian Patients Identified Through the MPS Brazil Network.
Mucopolysaccharidosis type IIIC (MPS IIIC) is a rare lysosomal storage disorder caused by pathogenic variants in the HGSNAT gene. Data from large patient cohorts remain scarce, particularly in Latin America. We retrospectively analyzed clinical, biochemical, and genetic data from patients diagnosed with MPS IIIC through the MPS Brazil Network. Diagnosis was based on reduced activity of acetyl-CoA:α-glucosaminide N-acetyltransferase (HGSNAT), elevated urinary glycosaminoglycans (uGAGs), and/or molecular genetics tests. A total of 101 patients were confirmed with MPS IIIC, representing one of the largest cohorts worldwide. Females accounted for 60% of cases. The mean age at symptom onset was 5.4 ± 3.9 years, while the mean age at diagnosis was 11.7 ± 6.9 years, reflecting a 6-year diagnostic delay. Most patients initially presented with developmental delay (82%) and facial dysmorphism (80%), whereas behavioral manifestations were less frequently identified (25%), suggesting a milder phenotype than previously reported. Genetic information was available for 28% of patients, showing recurrent alleles (c.372-2A>G, c.252dupT) and several novel mutations, which expand the mutational spectrum of the disease. Genotype-phenotype similarities with Portuguese, Italian, and Chinese cases suggest shared ancestry contributions. Regional differences included earlier diagnoses in the North of Brazil and high consanguinity rates in the Northeast region. This study describes the largest Brazilian cohort of MPS IIIC, documenting novel variants and regional heterogeneity. Findings highlight diagnostic delays, ancestry influences, and the urgent need for disease-modifying therapies.
Missing in action: the genetic mysteries of extremely low HDL cholesterol.
High-Density Lipoprotein Cholesterol (HDL-C) plays a pivotal role in cardiovascular health, acting as a key component in lipid transport and atheroprotection. While low HDL-C levels in the general population are often the result of multifactorial causes, extremely low HDL-C levels (<20 mg/dl) are rare and may be attributed to underlying genetic defects. Mutations in genes such as LCAT, APOA1, and ABCA1-although exceedingly rare-have been linked to profound alterations in lipid metabolism, often resulting in significant morbidity and increased cardiovascular risk. In this study, we used exome sequencing on patients with very low HDL-C. We identified three patients with pathogenic mutations associated with genetic low HDL-C syndrome, including ABCA1 [NM_005502.4(ABCA1):c.4175 + 1G > T, chr:9 91757308° C > A, rs375247413], LCAT [NM_000229.2(LCAT):c.349G > A p.Ala117Thr, rs28940886], and APOA1 [NM_000039.3(APOA1):c.388A > T, p.Lys130*]. Each case presented a unique spectrum of clinical phenotypes, systemic complications, and biochemical abnormalities, illustrating the diverse impact of these genetic mutations. We provide a detailed analysis of the clinical and biochemical profiles of these patients, highlighting key aspects of disease manifestation and progression. This report underscores the importance of recognizing and characterizing rare genetic causes of low HDL-C, which may have profound implications for patient care and risk stratification.
CCDC22 mutations that impair COMMD binding cause attenuated 3C/Ritscher-Schinzel syndrome.
The CCC complex, composed of CCDC22, CCDC93, and ten proteins of the COMMD family, coordinates several critical steps required to recycle internalized plasma membrane proteins from endosomes to the cell surface. CCC interacts with Retriever, a trimeric cargo recognition complex comprising VPS35L, VPS26C, and VPS29, and works closely with the WASH complex, a crucial regulator of branched actin polymerization at endosomal membranes. Mutations in genes encoding subunits of these three complexes, CCDC22, VPS35L, and WASHC5, have been linked with a developmental syndrome known as 3 C (cranio-cerebello-cardiac) or Ritscher-Schinzel syndrome. Here, we report a new CCDC22 missense mutation, p.E208K, that results in attenuated 3 C syndrome, without cardiac or neuroanatomical abnormalities. We show that this mutation impairs CCC complex assembly by disrupting a conserved interaction surface required for CCDC22-COMMD4 binding. We also review previously described cases and identify that CCDC22 p.P172R has a similar attenuated phenotype and impairs complex assembly in a similar fashion as p.E208K. The characterization of these mutations adds to our understanding of the clinical and molecular spectrum of these disorders.
Identification and characterization of novel genetic variants in the first Chinese family of mucopolysaccharidosis IIIC (Sanfilippo C syndrome).
Mucopolysaccharidosis type IIIC (MPS IIIC) is one of inherited lysosomal storage disorders, caused by deficiencies in lysosomal hydrolases degrading acidic mucopolysaccharides. The gene responsible for MPS IIIC is HGSNAT, which encodes an enzyme that catalyses the acetylation of the terminal glucosamine residues of heparan sulfate. So far, few studies have focused on the genetic landscape of MPS IIIC in China, where IIIA and IIIB were the major subtypes. In this study, we utilized whole-exome sequencing (WES) to identify novel compound heterozygous variants in the HGSNAT gene from a Chinese patient with typical MPS IIIC symptoms: c.743G>A; p.Gly248Glu and c.1030C>T; p.Arg344Cys. We performed in silico analysis and experimental validation, which confirmed the deleterious pathogenic nature of both variants, as evidenced by the loss of HGSNAT activity and failure of lysosomal localization. To the best of our knowledge, the MPS IIIC is first confirmed by clinical, biochemical and molecular genetic findings in China. Our study thus expands the spectrum of MPS IIIC pathogenic variants, which is of importance to dissect the pathogenesis and to carry out clinical diagnosis of MPS IIIC. Moreover, this study helps to depict the natural history of Chinese MPS IIIC populations.
Renal Involvement in Multisystem Inflammatory Syndrome in Children: Not Only Acute Kidney Injury.
Kidney involvement has been poorly investigated in SARS-CoV-2 Multisystem Inflammatory Syndrome in Children (MIS-C). To analyze the spectrum of renal involvement in MIS-C, we performed a single-center retrospective observational study including all MIS-C patients diagnosed at our Pediatric Department between April 2020 and May 2022. Demographic, clinical, pediatric intensive care unit (PICU) admission's need and laboratory data were collected at onset and after 6 months. Among 55 MIS-C patients enrolled in the study, kidney involvement was present in 20 (36.4%): 13 with acute kidney injury (AKI) and 7 with isolated tubular dysfunction (TD). In eight patients, concomitant AKI and TD was present (AKI-TD). AKI patients needed higher levels of intensive care (PICU: 61.5%, p < 0.001; inotropes: 46.2%, p = 0.002; second-line immuno-therapy: 53.8%, p < 0.001) and showed lower levels of HCO3- (p = 0.012), higher inflammatory markers [neutrophils (p = 0.092), PCT (p = 0.04), IL-6 (p = 0.007)] as compared to no-AKI. TD markers showed that isolated TD presented higher levels of HCO3- and lower inflammatory markers than AKI-TD. Our results indicate a combination of both pre-renal and inflammatory damage in the pathogenesis of kidney injury in MIS-C syndrome. We highlight, for the first time, the presence of tubular involvement in MIS-C, providing new insights in the evaluation of kidney involvement and its management in this condition.
Publicações recentes
Unveiling Mucopolysaccharidosis IIIC in Brazil: Diagnostic Journey and Clinical Features of Brazilian Patients Identified Through the MPS Brazil Network.
Missing in action: the genetic mysteries of extremely low HDL cholesterol.
CCDC22 mutations that impair COMMD binding cause attenuated 3C/Ritscher-Schinzel syndrome.
Identification and characterization of novel genetic variants in the first Chinese family of mucopolysaccharidosis IIIC (Sanfilippo C syndrome).
Renal Involvement in Multisystem Inflammatory Syndrome in Children: Not Only Acute Kidney Injury.
📚 EuropePMC33 artigos no totalmostrando 20
Unveiling Mucopolysaccharidosis IIIC in Brazil: Diagnostic Journey and Clinical Features of Brazilian Patients Identified Through the MPS Brazil Network.
Diseases (Basel, Switzerland)Missing in action: the genetic mysteries of extremely low HDL cholesterol.
Frontiers in cardiovascular medicineCCDC22 mutations that impair COMMD binding cause attenuated 3C/Ritscher-Schinzel syndrome.
BMC medical genomicsIdentification and characterization of novel genetic variants in the first Chinese family of mucopolysaccharidosis IIIC (Sanfilippo C syndrome).
Journal of cellular and molecular medicineRenal Involvement in Multisystem Inflammatory Syndrome in Children: Not Only Acute Kidney Injury.
Children (Basel, Switzerland)Case report: Exploring under the tip of the iceberg: A case series of "self-limiting" multisystem inflammatory syndrome in children.
Frontiers in pediatricsMIS-C: A COVID-19-as sociated condition between hypoimmunity and hyperimmunity.
Frontiers in immunologyCOVID-19 diversity: A case of multisystem inflammatory syndrome in children masquerading as juvenile systemic lupus erythematosus.
International journal of immunopathology and pharmacologyTargeted Genotyping of MIS-C Patients Reveals a Potential Alternative Pathway Mediated Complement Dysregulation during COVID-19 Infection.
Current issues in molecular biologyMechanisms of Immune Dysregulation in COVID-19 Are Different From SARS and MERS: A Perspective in Context of Kawasaki Disease and MIS-C.
Frontiers in pediatricsInvolvement of ADGRV1 Gene in Familial Forms of Genetic Generalized Epilepsy.
Frontiers in neurologyNeuronal and Astrocytic Differentiation from Sanfilippo C Syndrome iPSCs for Disease Modeling and Drug Development.
Journal of clinical medicineGeneration of two compound heterozygous HGSNAT-mutated lines from healthy induced pluripotent stem cells using CRISPR/Cas9 to model Sanfilippo C syndrome.
Stem cell researchA De Novo FOXP1 Truncating Mutation in a Patient Originally Diagnosed as C Syndrome.
Scientific reportsBohring-opitz syndrome - A case of a rare genetic disorder.
The Medical journal of MalaysiaA De Novo Nonsense Mutation in MAGEL2 in a Patient Initially Diagnosed as Opitz-C: Similarities Between Schaaf-Yang and Opitz-C Syndromes.
Scientific reportsCompound heterozygous mutations in the IFT140 gene cause Opitz trigonocephaly C syndrome in a patient with typical features of a ciliopathy.
Clinical geneticsScreening of CD96 and ASXL1 in 11 patients with Opitz C or Bohring-Opitz syndromes.
American journal of medical genetics. Part AActivity and High-Order Effective Connectivity Alterations in Sanfilippo C Patient-Specific Neuronal Networks.
Stem cell reportsEXTL2 and EXTL3 inhibition with siRNAs as a promising substrate reduction therapy for Sanfilippo C syndrome.
Scientific reportsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome C.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome C
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Unveiling Mucopolysaccharidosis IIIC in Brazil: Diagnostic Journey and Clinical Features of Brazilian Patients Identified Through the MPS Brazil Network.
- Missing in action: the genetic mysteries of extremely low HDL cholesterol.
- CCDC22 mutations that impair COMMD binding cause attenuated 3C/Ritscher-Schinzel syndrome.
- Identification and characterization of novel genetic variants in the first Chinese family of mucopolysaccharidosis IIIC (Sanfilippo C syndrome).
- Renal Involvement in Multisystem Inflammatory Syndrome in Children: Not Only Acute Kidney Injury.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1308(Orphanet)
- OMIM OMIM:211750(OMIM)
- MONDO:0008893(MONDO)
- GARD:5978(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q1022312(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome C
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Reposicionamento
- fonte: Drug Repurposing Hub