A hipertriptofanemia familiar é caracterizada por déficit intelectual associado a problemas comportamentais: alterações periódicas de humor, respostas afetivas exageradas e comportamento sexual anormal. Doze casos foram relatados até agora. Anormalidades congênitas no metabolismo do triptofano parecem ser responsáveis pela triptofanemia e triptofanúria.
Introdução
O que você precisa saber de cara
A hipertriptofanemia familiar é caracterizada por déficit intelectual associado a problemas comportamentais: alterações periódicas de humor, respostas afetivas exageradas e comportamento sexual anormal. Doze casos foram relatados até agora. Anormalidades congênitas no metabolismo do triptofano parecem ser responsáveis pela triptofanemia e triptofanúria.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 7 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 23 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Heme-dependent dioxygenase that catalyzes the oxidative cleavage of the L-tryptophan (L-Trp) pyrrole ring and converts L-tryptophan to N-formyl-L-kynurenine. Catalyzes the oxidative cleavage of the indole moiety
Hypertryptophanemia
An autosomal recessive condition characterized by persistent hypertryptophanemia and hyperserotoninemia.
Variantes genéticas (ClinVar)
33 variantes patogênicas registradas no ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Hipertriptofanemia
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Initiation of Brain Extract Fibrillation and Effective Cellular Internalization of Tryptophan Fibrils Unveils Its Neurotoxicity Risk.
Recent discoveries on the self-assembly of aromatic amino acids into amyloid-like neurotoxic nanostructures have initiated a quest to decode the molecular mechanisms for the initiation of neurodegeneration. Moreover, the multicomponent nature of the amyloid deposits still questions the existing and well-defined amyloid cascade hypothesis. Hence, deciphering the neurotoxicity of amyloid-like nanostructures of aromatic amino acids becomes crucial for understanding the etiology of amyloidogenesis. Here, we demonstrate the cellular internalization and consequential damaging effects of self-assembled amyloid-like tryptophan nanostructures on human neuroblastoma cells. The cell-damaging potential of tryptophan nanostructure seems to be facilitated via ROS generation, necrosis and apoptosis mediated cell death. Further, tryptophan nanostructures were found to be seeding competent conformers, which triggered aggressive aggregation of brain extract components. The early stage intermediate nanostructures possess a higher cross-seeding efficacy than the seeding potential of the matured tryptophan fibrils. In addition to the cell-damaging and cross-seeding effects, tryptophan fibrils were found to catalyze oxidation of neuromodulator dopamine. These findings add more insights into the specific role of tryptophan self-assembly during the pathogenesis of hypertryptophanemia and other amyloid-associated neurodegenerative complications.
Systemic tryptophan homeostasis.
Tryptophan is an essential amino acid, which is not only a building block for protein synthesis, but also a precursor for the biosynthesis of co-enzymes and neuromodulators, such as NAD/NADP(H), kynurenic acid, melatonin and serotonin. It also plays a role in immune homeostasis, as local tryptophan catabolism impairs T-lymphocyte mediated immunity. Therefore, tryptophan plasmatic concentration needs to be stable, in spite of large variations in dietary supply. Here, we review the main checkpoints accounting for tryptophan homeostasis, including absorption, transport, metabolism and elimination, and we discuss the physiopathology of disorders associated with their dysfunction. Tryptophan is catabolized along the kynurenine pathway through the action of two enzymes that mediate the first and rate-limiting step of the pathway: indoleamine 2,3-dioxygenase 1 (IDO1) and tryptophan 2,3-dioxygenase (TDO). While IDO1 expression is restricted to peripheral sites of immune modulation, TDO is massively expressed in the liver and accounts for 90% of tryptophan catabolism. Recent data indicated that the stability of the TDO protein is regulated by tryptophan and that this regulation allows a tight control of tryptophanemia. TDO is stabilized when tryptophan is abundant in the plasma, resulting in rapid degradation of dietary tryptophan. In contrast, when tryptophan is scarce, TDO is degraded by the proteasome to avoid excessive tryptophan catabolism. This is triggered by the unmasking of a degron in a non-catalytic tryptophan-binding site, resulting in TDO ubiquitination by E3 ligase SKP1-CUL1-F-box. Deficiency in TDO or in the hepatic aromatic transporter SLC16A10 leads to severe hypertryptophanemia, which can disturb immune and neurological homeostasis.
Tryptophan self-assembly yields cytotoxic nanofibers containing amyloid-mimicking and cross-seeding competent conformers.
Dietary consumption of Trp via protein-based foods is essential for the maintenance of crucial metabolic processes including the synthesis of proteins and several vital metabolites such as serotonin, melatonin, acetyl CoA, and NADP. However, the abnormal build-up of Trp is known to cause familial hypertryptophanemia and several brain-related medical complications. The molecular mechanism of the onset of such Trp-driven health issues is largely unknown. Here, we show that Trp, under the physiologically mimicked conditions of temperature and buffer, undergoes a concentration driven self-assembly process, yielding amyloid-mimicking nanofibers. Viable H-bonds, π-π interactions and hydrophobic contacts between optimally coordinated Trp molecules become important factors for the formation of a Trp nanoassembly that displays a hydrophobic exterior and a hydrophilic interior. Importantly, Trp nanofibers were found to possess high affinity for native proteins, and they act as cross-seeding competent conformers capable of nucleating amyloid formation in globular proteins including whey protein β-lactoglobulin and type II diabetes linked insulin hormone. Moreover, these amyloid mimicking Trp nanostructures showed toxic effects on neuroblastoma cells. Since the key symptoms in hypertryptophanemia such as behavioural defects and brain-damaging oxidative stress are also observed in amyloid related disorders, our findings on amyloid-like Trp-nanofibers may help in the mechanistic understanding of Trp-related complications and these findings are equally important for innovation in applied nanomaterials design and strategies.
Unusual Aggregates Formed by the Self-Assembly of Proline, Hydroxyproline, and Lysine.
There is a plethora of significant research that illustrates toxic self-assemblies formed by the aggregation of single amino acids, such as phenylalanine, tyrosine, tryptophan, cysteine, and methionine, and their implication on the etiology of inborn errors of metabolisms (IEMs), such as phenylketonuria, tyrosinemia, hypertryptophanemia, cystinuria, and hypermethioninemia, respectively. Hence, studying the aggregation behavior of single amino acids is very crucial from the chemical neuroscience perspective to understanding the common etiology between single amino acid metabolite disorders and amyloid diseases like Alzheimer's and Parkinson's. Herein we report the aggregation properties of nonaromatic single amino acids l-proline (Pro), l-hydroxyproline (Hyp), and l-lysine hydrochloride (Lys). The morphologies of the self-assembled structures formed by Pro, Hyp, and Lys were extensively studied by various microscopic techniques, and controlled morphological transitions were observed under varied concentrations and aging times. The mechanism of structure formation was deciphered by concentration-dependent 1H NMR analysis, which revealed the crucial role of hydrogen bonding and hydrophobic interactions in the structure formation of Pro, Hyp, and Lys. MTT assays on neural (SHSY5Y) cell lines revealed that aggregates formed by Pro, Hyp, and Lys reduced cell viability in a dose-dependent manner. These results may have important implications in the understanding of the patho-physiology of disorders such as hyperprolinemia, hyperhydroxyprolinemia, and hyperlysinemia since all these IEMs are associated with severe neurodegenerative symptoms, including intellectual disability, seizures, and psychiatric problems. Our future studies will endeavor to study these biomolecular assemblies in greater detail by immuno-histochemical analysis and advanced biophysical assays.
Hypertryptasemia and Mast Cell-Related Disorders in Severe Osteoporotic Patients.
Systemic mastocytosis (SM) is characterized by a clonal proliferation of neoplastic mast cells (MCs) in one or more extracutaneous organs including the bone marrow (BM). SM is often associated with osteoporosis (OP) and fractures. Hypertryptasemia usually occurs in SM. We investigated the prevalence of hypertryptasemia in a series of severe osteoporotic patients, the performance of the tryptase test in diagnosing SM in these patients, and their bone features. The medical records of 232 patients (168 females and 64 males) with a diagnosis of OP (50.4% with fractures) and a serum tryptase assessment were reviewed. BM assessment was performed in a subset of hypertryptasemic patients; clinical, biochemical, and radiographic data were collected. Hypertryptasemia was detected in 33 patients. BM assessment (n = 16) was normal in 8 hypertryptasemic patients, while BM criteria for the diagnosis of SM were met in 3 patients, MC alterations were detected in 4 patients, and one patient presented a polycythemia vera. Serum tryptase levels were higher than 11.4 ng/ml in all patients with BM alterations. The best cut-off of tryptase level related to BM alterations was 17.9 ng/ml, with a sensibility and sensitivity of 75% (AUC = 0.797 and P = 0.015 by ROC analysis). All osteoporotic patients with hypertryptasemia experienced at least one vertebral fracture associated with a severe reduction of the lumbar bone mineral density. The prevalence of MC-related disorders in severe OP was 3.0%, accounting for the 7.4% of the secondary causes of OP. MC-related disorders may be involved in bone fragility and assessment of serum tryptase is useful to detect MC-related disorders.
Publicações recentes
Initiation of Brain Extract Fibrillation and Effective Cellular Internalization of Tryptophan Fibrils Unveils Its Neurotoxicity Risk.
Tryptophan self-assembly yields cytotoxic nanofibers containing amyloid-mimicking and cross-seeding competent conformers.
🥇 Revisão sistemáticaSystemic tryptophan homeostasis.
Unusual Aggregates Formed by the Self-Assembly of Proline, Hydroxyproline, and Lysine.
Hypertryptophanemia due to tryptophan 2,3-dioxygenase deficiency.
📚 EuropePMC5 artigos no totalmostrando 10
Initiation of Brain Extract Fibrillation and Effective Cellular Internalization of Tryptophan Fibrils Unveils Its Neurotoxicity Risk.
ACS chemical neuroscienceTryptophan self-assembly yields cytotoxic nanofibers containing amyloid-mimicking and cross-seeding competent conformers.
NanoscaleSystemic tryptophan homeostasis.
Frontiers in molecular biosciencesUnusual Aggregates Formed by the Self-Assembly of Proline, Hydroxyproline, and Lysine.
ACS chemical neuroscienceHypertryptasemia and Mast Cell-Related Disorders in Severe Osteoporotic Patients.
Mediators of inflammationRationale and design of the Pemafibrate to Reduce Cardiovascular Outcomes by Reducing Triglycerides in Patients with Diabetes (PROMINENT) study.
American heart journalThe emerging role of novel antihyperglycemic agents in the treatment of heart failure and diabetes: A focus on cardiorenal outcomes.
Clinical cardiologyPrevalence of Positive Troponin and Echocardiogram Findings and Association With Mortality in Acute Ischemic Stroke.
StrokeHypertryptophanemia due to tryptophan 2,3-dioxygenase deficiency.
Molecular genetics and metabolismIs triglyceride therapy worth the effort?
Cleveland Clinic journal of medicineAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Hipertriptofanemia.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Hipertriptofanemia
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Initiation of Brain Extract Fibrillation and Effective Cellular Internalization of Tryptophan Fibrils Unveils Its Neurotoxicity Risk.
- Systemic tryptophan homeostasis.
- Tryptophan self-assembly yields cytotoxic nanofibers containing amyloid-mimicking and cross-seeding competent conformers.
- Unusual Aggregates Formed by the Self-Assembly of Proline, Hydroxyproline, and Lysine.
- Hypertryptasemia and Mast Cell-Related Disorders in Severe Osteoporotic Patients.
- Hypertryptophanemia due to tryptophan 2,3-dioxygenase deficiency.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2224(Orphanet)
- OMIM OMIM:600627(OMIM)
- MONDO:0010907(MONDO)
- GARD:2871(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q5958803(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
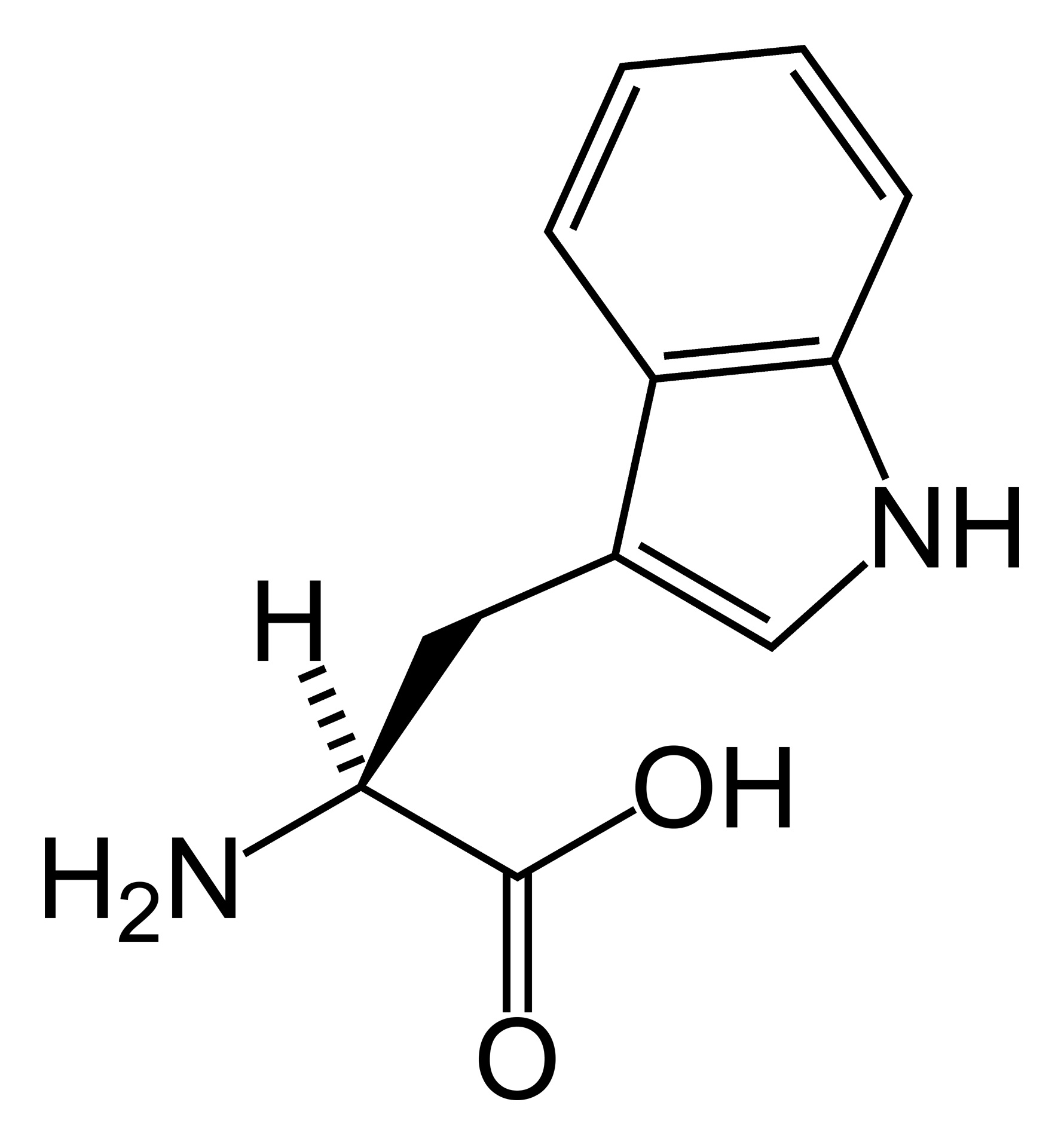
Hipertriptofanemia
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Reposicionamento
- fonte: Drug Repurposing Hub