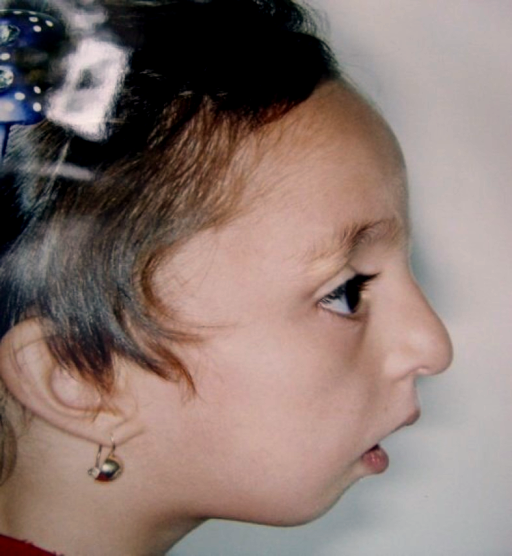
A Síndrome de Disostose Acrofacial tipo Rodriguez é uma síndrome com múltiplas malformações, na qual defeitos no desenvolvimento do rosto e da mandíbula, juntamente com deformidades graves de encurtamento ou ausência de membros, estão associados a malformações complexas de diferentes órgãos e sistemas. Isso inclui especialmente o sistema nervoso central (cérebro e medula espinhal), o trato urinário e reprodutor, o coração e os pulmões. O defeito na mandíbula e no rosto, caracterizado por uma mandíbula extremamente pequena e recuada (microretrognatismo grave) e fenda palatina (céu da boca aberto), causa a morte por dificuldade respiratória. A redução dos membros é grave e inclui o desenvolvimento incompleto dos ombros e da pelve, focomelia (membros muito curtos, com as mãos e os pés próximos ao tronco) com úmero (osso do braço) pouco desenvolvido, ausência dos ossos do antebraço (rádio e ulna), ausência completa dos ossos longos das pernas e várias anomalias nas mãos, predominantemente redução pré-axial (ausência dos polegares). Outras características incluem malformações do sistema nervoso central (como ausência do corpo caloso, estrutura que conecta os dois lados do cérebro, e estreitamento do aqueduto cerebral, um canal no cérebro), anomalias pulmonares (como ausência da divisão natural dos pulmões em lobos), malformações cardíacas complexas e útero unicorno (um útero com apenas um lado desenvolvido). Esses bebês também apresentam características faciais atípicas e anomalias nas orelhas. É uma condição rara, herdada de forma autossômica recessiva (o que significa que ambos os pais precisam ser portadores do gene para que o bebê desenvolva a síndrome). O prognóstico é desfavorável, e essa condição leva à morte ainda no útero ou logo após o nascimento.
Introdução
O que você precisa saber de cara
A Síndrome de Disostose Acrofacial tipo Rodriguez é uma síndrome com múltiplas malformações, na qual defeitos no desenvolvimento do rosto e da mandíbula, juntamente com deformidades graves de encurtamento ou ausência de membros, estão associados a malformações complexas de diferentes órgãos e sistemas. Isso inclui especialmente o sistema nervoso central (cérebro e medula espinhal), o trato urinário e reprodutor, o coração e os pulmões. O defeito na mandíbula e no rosto, caracterizado por uma mandíbula extremamente pequena e recuada (microretrognatismo grave) e fenda palatina (céu da boca aberto), causa a morte por dificuldade respiratória. A redução dos membros é grave e inclui o desenvolvimento incompleto dos ombros e da pelve, focomelia (membros muito curtos, com as mãos e os pés próximos ao tronco) com úmero (osso do braço) pouco desenvolvido, ausência dos ossos do antebraço (rádio e ulna), ausência completa dos ossos longos das pernas e várias anomalias nas mãos, predominantemente redução pré-axial (ausência dos polegares). Outras características incluem malformações do sistema nervoso central (como ausência do corpo caloso, estrutura que conecta os dois lados do cérebro, e estreitamento do aqueduto cerebral, um canal no cérebro), anomalias pulmonares (como ausência da divisão natural dos pulmões em lobos), malformações cardíacas complexas e útero unicorno (um útero com apenas um lado desenvolvido). Esses bebês também apresentam características faciais atípicas e anomalias nas orelhas. É uma condição rara, herdada de forma autossômica recessiva (o que significa que ambos os pais precisam ser portadores do gene para que o bebê desenvolva a síndrome). O prognóstico é desfavorável, e essa condição leva à morte ainda no útero ou logo após o nascimento.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 15 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 43 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive.
Component of the 17S U2 SnRNP complex of the spliceosome, a large ribonucleoprotein complex that removes introns from transcribed pre-mRNAs (PubMed:10882114, PubMed:12234937, PubMed:27720643, PubMed:32494006). The 17S U2 SnRNP complex (1) directly participates in early spliceosome assembly and (2) mediates recognition of the intron branch site during pre-mRNA splicing by promoting the selection of the pre-mRNA branch-site adenosine, the nucleophile for the first step of splicing (PubMed:12234937
Nucleus
Acrofacial dysostosis 1, Nager type
A form of acrofacial dysostosis, a group of disorders which are characterized by malformation of the craniofacial skeleton and the limbs. The major facial features of AFD1 include downslanted palpebral fissures, midface retrusion, and micrognathia, the latter of which often requires the placement of a tracheostomy in early childhood. Limb defects typically involve the anterior (radial) elements of the upper limbs and manifest as small or absent thumbs, triphalangeal thumbs, radial hyoplasia or aplasia, and radioulnar synostosis. Phocomelia of the upper limbs and, occasionally, lower-limb defects have also been reported.
Variantes genéticas (ClinVar)
69 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
4 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Disostose acro-facial, tipo Rodriguez
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Human stem cell model of neural crest cell differentiation reveals a requirement of SF3B4 in survival, maintenance, and differentiation.
In vitro modeling is a powerful approach to investigate the pathomechanisms driving human congenital conditions. Here, we use human embryonic stem cells (hESCs) to model Nager and Rodriguez syndromes, two craniofacial conditions characterized by hypoplastic neural crest-derived craniofacial bones, caused by pathogenic variants of SF3B4, a core component of the spliceosome. We observed that siRNA-mediated knockdown of SF3B4 interferes with the production of hESC-derived neural crest cells, as seen by a marked reduction in neural crest gene expression. This phenotype is associated with an increase in neural crest cell apoptosis and premature neuronal differentiation. Altogether, these results point to a role of SF3B4 in neural crest cell survival, maintenance, and differentiation. We propose that the dysregulation of these processes may contribute to Nager/Rodriguez syndrome-associated craniofacial defects.
The final demise of Rodriguez lethal acrofacial dysostosis: A case report and review of the literature.
We evaluated a newborn with acrofacial dysostosis in whom a clinical diagnosis of Nager syndrome was entertained. Radiographs revealed hypoplasia of the scapulae and bilateral humeroradial synostosis, with absent ulna on the left and hypoplastic ulna on the right. The finding of bilateral humeroradial synostosis had not been seen in cases of Nager syndrome before and we considered other diagnoses. Humeroradial synostosis has been found in three cases of acrofacial dysostosis Rodriguez type, a syndrome characterized by mandibular hypoplasia, upper and lower extremity phocomelia, and oligodactyly of the upper limbs. More recently, haploinsufficiency of the SF3B4 gene has been identified as the cause of both Nager and Rodriguez syndrome, leading many to believe that Rodriguez syndrome represents a more severe end of a Nager syndrome spectrum. An SF3B4 mutation was found in our patient, prompting a review of the previous known cases of Rodriguez syndrome, which revealed no clustering of SF3B4 mutations, and four cases of Rodriguez syndrome with mutations identical to those in cases of Nager syndrome. Rodriguez syndrome was previously thought of as a lethal acrofacial dysostosis distinct from Nager syndrome. A number of more mild cases, as well as our case, intermediate between the two phenotypes, illustrate that Rodriguez syndrome is a severe manifestation of Nager syndrome, and is not lethal with aggressive medical care.
Altered mRNA Splicing, Chondrocyte Gene Expression and Abnormal Skeletal Development due to SF3B4 Mutations in Rodriguez Acrofacial Dysostosis.
The acrofacial dysostoses (AFD) are a genetically heterogeneous group of inherited disorders with craniofacial and limb abnormalities. Rodriguez syndrome is a severe, usually perinatal lethal AFD, characterized by severe retrognathia, oligodactyly and lower limb abnormalities. Rodriguez syndrome has been proposed to be a severe form of Nager syndrome, a non-lethal AFD that results from mutations in SF3B4, a component of the U2 small nuclear ribonucleoprotein particle (U2 snRNP). Furthermore, a case with a phenotype intermediate between Rodriguez and Nager syndromes has been shown to have an SF3B4 mutation. We identified heterozygosity for SF3B4 mutations in Rodriguez syndrome, confirming that the phenotype is a dominant disorder that is allelic with Nager syndrome. The mutations led to reduced SF3B4 synthesis and defects in mRNA splicing, primarily exon skipping. The mutations also led to reduced expression in growth plate chondrocytes of target genes, including the DLX5, DLX6, SOX9, and SOX6 transcription factor genes, which are known to be important for skeletal development. These data provide mechanistic insight toward understanding how SF3B4 mutations lead to the skeletal abnormalities observed in the acrofacial dysostoses.
Rodriguez acrofacial dysostosis is caused by apparently de novo heterozygous mutations in the SF3B4 gene.
Acrofacial dysostosis syndrome of Rodriguez is characterized by severe mandibular underdevelopment, upper limb phocomelia with absent fingers, absent fibulae, cleft palate, microtia, and abnormal pulmonary function. First reported in three siblings it was assumed to be an autosomal recessive condition. However, subsequent publication reported a further five simplex occurrences and a living patient with a heterozygous mutation in the SF3B4 gene. Exome sequencing was performed on four fetuses with this disorder, including one of the originally described affected siblings. We identified two heterozygous frameshift mutations in the SF3B4 gene in three of the four fetuses investigated. The observed mutation was apparently de novo in one fetus for whom parental DNA was available. Thus, Acrofacial dysostosis syndrome of Rodriguez is an autosomal dominant condition and the recurrences identified in the initial report were likely due to gonadal mosaicism. © 2016 Wiley Periodicals, Inc.
Rodriguez lethal acrofacial dysostosis syndrome with ambiguous genitalia.
Publicações recentes
Ver todas no PubMed📚 EuropePMCmostrando 5
Human stem cell model of neural crest cell differentiation reveals a requirement of SF3B4 in survival, maintenance, and differentiation.
Developmental dynamics : an official publication of the American Association of AnatomistsThe final demise of Rodriguez lethal acrofacial dysostosis: A case report and review of the literature.
American journal of medical genetics. Part ARodriguez acrofacial dysostosis is caused by apparently de novo heterozygous mutations in the SF3B4 gene.
American journal of medical genetics. Part AAltered mRNA Splicing, Chondrocyte Gene Expression and Abnormal Skeletal Development due to SF3B4 Mutations in Rodriguez Acrofacial Dysostosis.
PLoS geneticsRodriguez lethal acrofacial dysostosis syndrome with ambiguous genitalia.
Taiwanese journal of obstetrics & gynecologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Disostose acro-facial, tipo Rodriguez.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Disostose acro-facial, tipo Rodriguez
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Human stem cell model of neural crest cell differentiation reveals a requirement of SF3B4 in survival, maintenance, and differentiation.Developmental dynamics : an official publication of the American Association of Anatomists· 2025· PMID 40047147mais citado
- The final demise of Rodriguez lethal acrofacial dysostosis: A case report and review of the literature.
- Altered mRNA Splicing, Chondrocyte Gene Expression and Abnormal Skeletal Development due to SF3B4 Mutations in Rodriguez Acrofacial Dysostosis.
- Rodriguez acrofacial dysostosis is caused by apparently de novo heterozygous mutations in the SF3B4 gene.
- Rodriguez lethal acrofacial dysostosis syndrome with ambiguous genitalia.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1788(Orphanet)
- OMIM OMIM:201170(OMIM)
- MONDO:0008714(MONDO)
- GARD:496(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q21154051(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Disostose acro-facial, tipo Rodriguez
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata