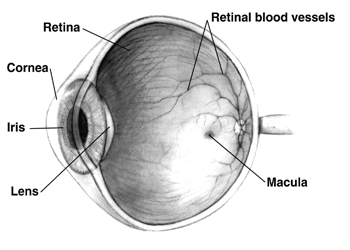
A hipoplasia macular é uma condição médica rara que envolve o subdesenvolvimento da mácula, uma pequena área na retina responsável pela visão em detalhes e pela percepção de luz. A hipoplasia macular está frequentemente associada ao albinismo.
Introdução
O que você precisa saber de cara
Visão geral
A Síndrome de hipoplasia da fóvea-catarata pré-senil é uma doença genética rara que afeta principalmente os olhos. A condição é caracterizada pelo desenvolvimento incompleto da fóvea (hipoplasia da fóvea), que é a região central da retina responsável pela visão de detalhes, e pelo aparecimento precoce de catarata (catarata pré-senil), geralmente na idade adulta. A prevalência estimada é de menos de 1 caso por 1.000.000 de pessoas.[1][3]
Sinais e sintomas
Os principais sinais e sintomas incluem: anormalidades da visão (como baixa acuidade visual), nistagmo (movimentos involuntários dos olhos), estrabismo (desalinhamento ocular), catarata (opacificação do cristalino) e atrofia óptica (dano ao nervo óptico). Além disso, pode haver hiperpigmentação generalizada da pele. Os sintomas geralmente se manifestam na idade adulta.[1][3]
Causas genéticas
Diagnóstico
Tratamento e manejo
Não há cura para a síndrome. O manejo é focado no tratamento dos sintomas, como a correção cirúrgica da catarata e o acompanhamento oftalmológico regular para monitorar a acuidade visual e possíveis complicações. Não existem medicamentos específicos aprovados para a doença. No Brasil, a condição não possui cobertura pelo Sistema Único de Saúde (SUS) para procedimentos específicos listados.[1][5]
Tratamentos citados na literatura
A literatura científica menciona algumas substâncias em associação com a doença, mas isso não representa recomendação de tratamento. As substâncias e o número de publicações encontradas são: Gadolinium DTPA (3), Infliximab (3), Mustard Gas (2), Cortisone (2), Tacrolimus (2), Oxygen (1), 1 Butanol (1), 3' sialyllactose (1), 3 IB MECA (1) e Alcohols (1). Esses dados foram minerados da base PubTator3 e não indicam eficácia ou segurança para uso clínico.[5]
Prognóstico e qualidade de vida
O prognóstico varia conforme a gravidade dos sintomas. A catarata pré-senil pode ser tratada cirurgicamente, mas a hipoplasia da fóvea e a atrofia óptica podem resultar em comprometimento visual permanente. O acompanhamento multidisciplinar com oftalmologista e geneticista é importante para o manejo adequado e suporte ao paciente.[1][3]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
A hipoplasia macular é uma condição médica rara que envolve o subdesenvolvimento da mácula, uma pequena área na retina responsável pela visão em detalhes e pela percepção de luz. A hipoplasia macular está frequentemente associada ao albinismo.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Síndrome de hipoplasia da fóvea-catarata pré-senil é uma doença genética rara que afeta principalmente os olhos. A condição é caracterizada pelo desenvolvimento incompleto da fóvea (hipoplasia da fóvea), que é a região central da retina responsável pela visão de detalhes, e pelo aparecimento precoce de catarata (catarata pré-senil), geralmente na idade adulta. A prevalência estimada é de menos de 1 caso por 1.000.000 de pessoas.[1][3]
Sinais e sintomas
Os principais sinais e sintomas incluem: anormalidades da visão (como baixa acuidade visual), nistagmo (movimentos involuntários dos olhos), estrabismo (desalinhamento ocular), catarata (opacificação do cristalino) e atrofia óptica (dano ao nervo óptico). Além disso, pode haver hiperpigmentação generalizada da pele. Os sintomas geralmente se manifestam na idade adulta.[1][3]
Causas genéticas
Diagnóstico
Tratamento e manejo
Não há cura para a síndrome. O manejo é focado no tratamento dos sintomas, como a correção cirúrgica da catarata e o acompanhamento oftalmológico regular para monitorar a acuidade visual e possíveis complicações. Não existem medicamentos específicos aprovados para a doença. No Brasil, a condição não possui cobertura pelo Sistema Único de Saúde (SUS) para procedimentos específicos listados.[1][5]
Tratamentos citados na literatura
A literatura científica menciona algumas substâncias em associação com a doença, mas isso não representa recomendação de tratamento. As substâncias e o número de publicações encontradas são: Gadolinium DTPA (3), Infliximab (3), Mustard Gas (2), Cortisone (2), Tacrolimus (2), Oxygen (1), 1 Butanol (1), 3' sialyllactose (1), 3 IB MECA (1) e Alcohols (1). Esses dados foram minerados da base PubTator3 e não indicam eficácia ou segurança para uso clínico.[5]
Prognóstico e qualidade de vida
O prognóstico varia conforme a gravidade dos sintomas. A catarata pré-senil pode ser tratada cirurgicamente, mas a hipoplasia da fóvea e a atrofia óptica podem resultar em comprometimento visual permanente. O acompanhamento multidisciplinar com oftalmologista e geneticista é importante para o manejo adequado e suporte ao paciente.[1][3]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 1 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 7 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Transcription factor with important functions in the development of the eye, nose, central nervous system and pancreas. Required for the differentiation of pancreatic islet alpha cells (By similarity). Competes with PAX4 in binding to a common element in the glucagon, insulin and somatostatin promoters. Regulates specification of the ventral neuron subtypes by establishing the correct progenitor domains (By similarity). Acts as a transcriptional repressor of NFATC1-mediated gene expression (By s
Nucleus
Aniridia 1
A congenital, bilateral, panocular disorder characterized by complete absence of the iris or extreme iris hypoplasia. Aniridia is not just an isolated defect in iris development but it is associated with macular and optic nerve hypoplasia, cataract, corneal changes, nystagmus. Visual acuity is generally low but is unrelated to the degree of iris hypoplasia. Glaucoma is a secondary problem causing additional visual loss over time.
Variantes genéticas (ClinVar)
543 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
5 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de hipoplasia da fóvea-catarata pré-senil
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Exploring Concomitant Ophthalmic Comorbidities in Portuguese Patients with Inherited Retinal Diseases: A Comprehensive Clinical Study.
Background/Objectives: Inherited retinal diseases (IRDs) are a heterogeneous group of rare eye disorders characterized by progressive photoreceptor degeneration, leading to severe visual impairment or even blindness. This study aims to investigate the prevalence, types, and clinical significance of ophthalmic comorbidities in Portuguese patients with IRDs. Methods: This nationwide Portuguese population-based retrospective study was based on the IRD-PT registry (retina.com.pt). Statistical analysis was conducted using Microsoft® Excel® for Microsoft 365 and IBM SPSS Statistics version 29.0.2.0. Informed consent was obtained from all participants. Results: A total of 1531 patients (1254 families) from six centers were enrolled. The cohort consisted of 51% males, with a mean age of 45.8 ± 19.3 years and a mean age at diagnosis of 39.4 ± 19.5 years. Overall, ocular comorbidities were reported in 644 patients (42.1%). In 176 individuals (11.5%), multiple concurrent comorbidities were found. Cataract was the most common comorbidity (21.3%), followed by amblyopia (6.3%) and high myopia (5.9%). Statistically significant associations with ocular comorbidities were observed in isolated progressive IRDs. Specifically, AR RP was associated with cataract (p < 0.001), and gene analysis revealed several significant associations. CRB1 was statistically linked to epiretinal membrane (ERM) (p = 0.003), EYS with cataract (p = 0.001), PROM1 with choroidal neovascularization (CNV) (p = 0.0026), and USH2A with macular hole (p = 0.01). Patients with the RPE65 mutation in Leber congenital amaurosis were associated with ERM (p = 0.019). There was also a significant association between X-linked RP and high myopia (p < 0.001) and CNV in Best disease (p < 0.001); in syndromic IRDs, cataract, cystoid macular edema, and ERM were observed in Usher syndrome, p = 0.002, p = 0.002, and p = 0.005, respectively, and the MYO7A gene was linked to cataract (p = 0.041) and strabismus (p = 0.013); pseudoxanthoma elasticum was significantly associated with CNV (p = 0.002); and foveal hypoplasia was associated with anterior segment dysgenesis (p < 0.001). Conclusions: This study enhances the current understanding of ocular comorbidities in IRDs in Portuguese patients. Common findings were cataract, refractive error, and CME. Stationary IRDs and pattern dystrophies showed fewer concomitant comorbidities, supporting their classification as non-progressive or benign conditions. The significance of registries like IRD-PT cannot be overstated, particularly in the context of rare diseases. These databases serve multiple crucial functions in enabling detailed documentation of disease characteristics and long-term monitoring of disease progression.
Foveal hypoplasia in Myhre syndrome: a novel association.
Myhre syndrome is an autosomal dominant condition caused by pathogenic variants in the transcriptional co-regulator SMAD4. Myhre syndrome is characterized by distinctive facial features, short stature, musculoskeletal abnormalities, and intellectual disability. Reported ocular abnormalities include refractive errors, corectopia, cataract, strabismus, and pseudo) papilledema. We describe an 8-year-old boy with Myhre syndrome due to a c.1498A > G; p.I500V pathogenic variant in SMAD4. Ocular examination revealed bilateral emmetropia, mild visual acuity reduction in the right eye (20/25), grade 1b foveal hypoplasia in both eyes and small optic discs with pseudopapilledema. This report marks the first reported case of foveal hypoplasia in Myhre syndrome, a potentially underreported finding, given the relative lack of OCT assessment in patients with Myhre syndrome. We discuss pathophysiological link between foveal hypoplasia and gain-of-function variants in SMAD4.
Detailed structural abnormalities associated with a novel VCAN variant in a family with versican vitreoretinopathy.
To understand the retina micropathology in a family with a novel variant in VCAN. Two sisters ages 16 (proband) and 18 years old and their 48-year-old father underwent comprehensive ophthalmic evaluations. Multimodal imaging was performed with spectral domain optical coherence tomography, ultrawide field short-wavelength fundus autofluorescence, and pseudocolor imaging. Cataracts were present in the sisters along with a penetrant retinal phenotype in all three patients with vitreoretinal ring opacities and traction, peripheral pigmentary clumps, lattice-like features, retinoschisis, foveal ectopia, and nasal displacement of vessels. There were regions with inner retinal thinning with spared outer retina, likely a consequence of vitreoretinal traction, that contrasted with large areas of profound photoreceptor degeneration, but with a rather normal or thickened inner retina. A previously unreported heterozygous variant in intron 7 of VCAN (c.4004-2A>C) segregated with the phenotype in the proband and her father. Segregation of a versican-associated vitreoretinopathy supports the pathogenicity of the VCAN variant. The patterns of structural abnormalities support classical mechanisms of disease that involve local vitreoretinal traction, as well as possible alternative developmental and/or degenerative changes of the retina, RPE, and/or choroid that result from the primary molecular defect.
Comprehensive Analysis of Congenital Aniridia and Differential Diagnoses: Genetic Insights and Clinical Manifestations.
Congenital aniridia (CA) is a severe and complex disorder involving the entire eye, primarily characterized by iris anomalies alongside other clinical features that pose significant risks to vision. This study seeks to offer a comprehensive overview of CA by detailing its clinical presentations, genetic underpinnings, associated phenotypes, and differential diagnoses. Additionally, it proposes a diagnostic framework to distinguish CA from other conditions that present with similar iris abnormalities. We conducted a comprehensive literature review to compile and analyze clinical and genetic data related to CA and its differential diagnoses. We included all studies describing the clinical characteristics, pathogenic variants, and associated syndromes of congenital aniridia. CA presents a wide range of ocular symptoms. Pathogenic variants in the PAX6 gene are the primary genetic cause of CA, though variations in other genes, including FOXC1, PITX2, CYP1B1, FOXD3, PITX3, CPAMD8, ITPR1, TENM3, TRIM44, COL4A1, CRYAA, and PXDN may also be implicated. The differential diagnosis of CA requires careful consideration of conditions with overlapping symptoms, such as WAGR syndrome (which involves deletions affecting the PAX6 and WT1 genes on chromosome 11p13, and potentially BDNF on 11p14.1), Axenfeld-Rieger syndrome (FOXC1/PITX2), ring-chromosome 6 syndrome (which involves FOXC1 microdeletion), COL4A1-related anterior segment dysgenesis, Gillespie syndrome (ITPR1 gene) or Peters anomaly. Accurate diagnosis can be achieved by evaluating specific clinical features-including iris anomalies, aniridia-associated keratopathy, cataracts, glaucoma, foveal hypoplasia, nystagmus, and optic nerve head abnormalities-supplemented by genetic testing. Understanding the diverse clinical presentations and genetic basis of diseases associated with iris abnormalities is essential for accurate diagnosis and effective management. Integrating genetic diagnostics into the evaluation process enables the development of tailored treatment strategies, which can significantly improve patient outcomes.
Novel compound heterozygous variants in SIX6 cause a PAX2 like Dysplastic Optic Disc with macular abnormalities without coexistent microphthalmia or cataract.
Congenital optic disc dysplasia with coexistent macular abnormalities is seen in Morning-glory disc anomaly, optic disc pit, PAX6-related disc coloboma, and in the PAX2-related Papillo-renal syndrome. We report novel compound heterozygous variants in SIX6 causing optic disc dysplasia and macular abnormality without coexisting cataract or microphthalmia for the first time in Chinese ethnicity and also describe the macular OCT findings. A 4 year-8 months-old female child presented with poor vision, photophobia, and nystagmus noticed 4 months after her birth was diagnosed elsewhere to have isolated cone dystrophy. She was evaluated subsequently by an ocular geneticist. She underwent a complete ophthalmic evaluation including orthoptic evaluation, cycloplegic refraction, and a dilated fundus evaluation. She had fundus photography, macular OCT, and ERG. She had systemic evaluations, relevant systemic investigations, and molecular genetic testing. Whole Exome Sequencing (WES) revealed compound heterozygous variants both in the SIX6 and in the TULP1 gene. No pathogenic variants were identified in the PAX2, PAX6 or in any of the other developmental genes or in the genes currently known to cause cone dystrophy or cone-rod dystrophy. Parents were heterozygous for variants in both SIX6 and TULP1. Homozygous or compound heterozygous pathogenic variants in SIX6 can cause a dysplastic optic disc and coexistent macular abnormalities. This dysplastic disc may be clinically indistinguishable from that seen in the PAX2 related Papillo-renal syndrome. Careful optic disc evaluation of subtle disc dysplasia is critical in differentiating this extremely rare entity from other relatively common causes of isolated cone dystrophies or cone-rod dystrophies.
Publicações recentes
An interconnected data infrastructure to support large-scale rare disease research.
A novel MT-ATP6 variant associated with complicated ataxia in two unrelated Italian patients: case report and functional studies.
Phenotypically Similar Rare Disease Identification from an Integrative Knowledge Graph for Data Harmonization: Preliminary Study.
📚 EuropePMCmostrando 35
Exploring Concomitant Ophthalmic Comorbidities in Portuguese Patients with Inherited Retinal Diseases: A Comprehensive Clinical Study.
GenesFoveal hypoplasia in Myhre syndrome: a novel association.
Ophthalmic geneticsDetailed structural abnormalities associated with a novel VCAN variant in a family with versican vitreoretinopathy.
Ophthalmic geneticsComprehensive Analysis of Congenital Aniridia and Differential Diagnoses: Genetic Insights and Clinical Manifestations.
Ophthalmology and therapyNovel compound heterozygous variants in SIX6 cause a PAX2 like Dysplastic Optic Disc with macular abnormalities without coexistent microphthalmia or cataract.
Ophthalmic geneticsVogt-Koyanagi-Harada-like disease secondary to anticancer treatment: a multicentre case series.
Eye (London, England)Multimodal Evaluation and Management of Wagner Syndrome-Three Patients from an Affected Family.
GenesFoveal photoreceptor atrophy, persistent fetal vasculature, congenital cataracts, and microphthalmia in a pediatric patient with BCOR-associated oculo-facio-cardio-dental (OFCD) syndrome.
American journal of ophthalmology case reportsCalciphylaxis, beware the ophthalmic mimic: A case series.
Clinical nephrology. Case studiesCorrelation between Severity of Idiopathic Epiretinal Membrane and Irvine-Gass Syndrome.
Journal of personalized medicineFoveal Hypoplasia Related to Congenital Rubella.
CureusWaardenburg syndrome-associated neurotrophic keratopathy in a child treated with neurotization surgery.
Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and StrabismusLong-term efficacy of dexamethasone intravitreal implant in the treatment of Vogt-Koyanagi-Harada disease relapsing posterior uveitis.
Indian journal of ophthalmology[National protocol for diagnosis and care of congenital aniridia: Summary for the attending physician].
Journal francais d'ophtalmologieFrequency of cystoid macular edema and vitreomacular interface disorders in genetically solved syndromic and non-syndromic retinitis pigmentosa.
Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle OphthalmologieSpontaneous closure of a chronic full-thickness idiopathic macular hole after Irvine-Gass syndrome resolution.
BMC ophthalmologyAxenfeld-Rieger syndrome combined with a foveal anomaly in a three-generation family: a case report.
BMC ophthalmologyDescriptive Study of a Cohort of 488 Patients with Inherited Retinal Dystrophies.
Clinical ophthalmology (Auckland, N.Z.)Management of cataract in a case of retinitis pigmentosa with bilateral pseudoexfoliation syndrome.
BMJ case reportsGlued intraocular lens in eyes with deficient capsules: retrospective analysis of long-term effects.
Journal of cataract and refractive surgeryAnti-interleukin-6 receptor therapy with tocilizumab for refractory pseudophakic cystoid macular edema.
American journal of ophthalmology case reportsNovel Intragenic PAX6 Deletion in a Pedigree with Aniridia, Morbid Obesity, and Diabetes.
Current eye research[Congenital aniridia in children].
La Revue du praticienTOXIC POSTERIOR SEGMENT SYNDROME AFTER DROPLESS CATARACT SURGERY WITH COMPOUNDED TRIAMCINOLONE-MOXIFLOXACIN.
Retina (Philadelphia, Pa.)Multimodal imaging in a patient with Prader-Willi syndrome.
International journal of retina and vitreousOptical coherence tomography features in a case of Type I sialidosis.
Taiwan journal of ophthalmologyNovel case of paternal paracentric inversion causing partial trisomy 13 and review of the literature.
American journal of medical genetics. Part A[Multimodal Approaches for the Analysis of Retinal Functional Disorders―Focusing on Retinal Detachment].
Nippon Ganka Gakkai zasshiTopical bromfenac for prevention and treatment of cystoid macular edema following cataract surgery: a review.
Clinical ophthalmology (Auckland, N.Z.)New truncation mutation of the NR2E3 gene in a Japanese patient with enhanced S-cone syndrome.
Japanese journal of ophthalmologyRetinal features in Mulvihill-Smith syndrome.
Ophthalmic geneticsClinical outcomes of double membrane peeling with or without simultaneous phacoemulsification/gas tamponade for vitreoretinal-interface-associated (VRI) disorders.
International ophthalmologyDexamethasone implant as an effective treatment option for macular edema due to Irvine-Gass syndrome.
Journal of cataract and refractive surgeryGPR143 Gene Mutations in Five Chinese Families with X-linked Congenital Nystagmus.
Scientific reportsAnirdia-like phenotype caused by 6p25 dosage aberrations.
American journal of medical genetics. Part AAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de hipoplasia da fóvea-catarata pré-senil.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de hipoplasia da fóvea-catarata pré-senil
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Exploring Concomitant Ophthalmic Comorbidities in Portuguese Patients with Inherited Retinal Diseases: A Comprehensive Clinical Study.
- Foveal hypoplasia in Myhre syndrome: a novel association.
- Detailed structural abnormalities associated with a novel VCAN variant in a family with versican vitreoretinopathy.
- Comprehensive Analysis of Congenital Aniridia and Differential Diagnoses: Genetic Insights and Clinical Manifestations.
- Novel compound heterozygous variants in SIX6 cause a PAX2 like Dysplastic Optic Disc with macular abnormalities without coexistent microphthalmia or cataract.
- An interconnected data infrastructure to support large-scale rare disease research.
- A novel MT-ATP6 variant associated with complicated ataxia in two unrelated Italian patients: case report and functional studies.
- Phenotypically Similar Rare Disease Identification from an Integrative Knowledge Graph for Data Harmonization: Preliminary Study.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2253(Orphanet)
- MONDO:0016395(MONDO)
- GARD:406(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q56013824(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome de hipoplasia da fóvea-catarata pré-senil
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata