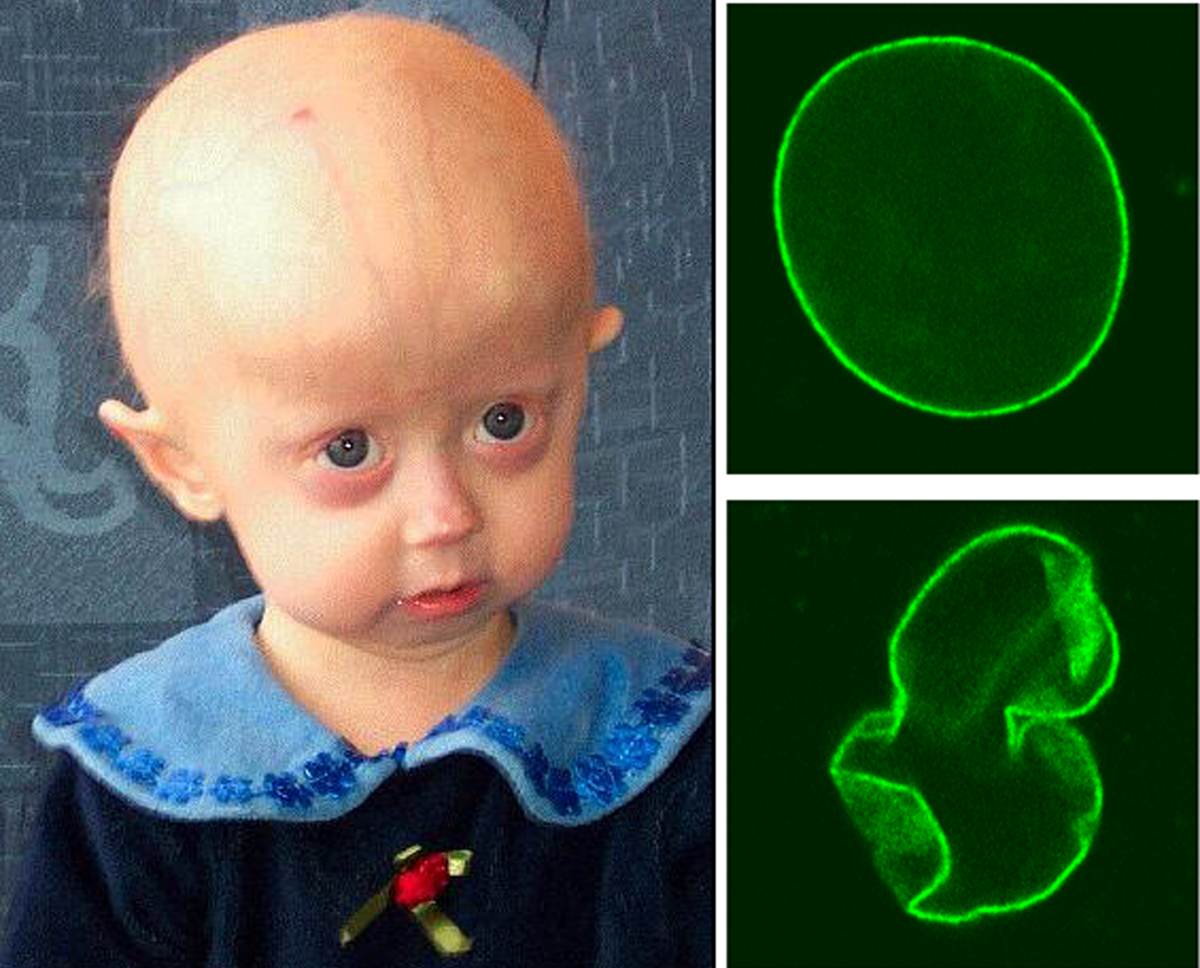
A Síndrome de Wiedemann-Rautenstrauch é uma condição muito rara com características de envelhecimento precoce que podem ser notadas ao nascer, pouca gordura debaixo da pele, poucos pelos e cabelos, cabeça com tamanho maior que o esperado e características físicas incomuns.
Introdução
O que você precisa saber de cara
A Síndrome de Wiedemann-Rautenstrauch é uma condição muito rara com características de envelhecimento precoce que podem ser notadas ao nascer, pouca gordura debaixo da pele, poucos pelos e cabelos, cabeça com tamanho maior que o esperado e características físicas incomuns.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 64 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 189 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisCatalytic core component of RNA polymerase III (Pol III), a DNA-dependent RNA polymerase which synthesizes small non-coding RNAs using the four ribonucleoside triphosphates as substrates. Synthesizes 5S rRNA, snRNAs, tRNAs and miRNAs from at least 500 distinct genomic loci (PubMed:19609254, PubMed:19631370, PubMed:20413673, PubMed:33335104, PubMed:33558764, PubMed:33558766, PubMed:34675218, PubMed:35637192, PubMed:9331371). Pol III-mediated transcription cycle proceeds through transcription init
NucleusCytoplasm, cytosol
Leukodystrophy, hypomyelinating, 7, with or without oligodontia and/or hypogonadotropic hypogonadism
An autosomal recessive neurodegenerative disorder characterized by childhood onset of progressive motor decline manifest as spasticity, ataxia, tremor, and cerebellar signs, as well as mild cognitive regression. Other features may include hypodontia or oligodontia and hypogonadotropic hypogonadism. There is considerable inter- and intrafamilial variability.
Variantes genéticas (ClinVar)
436 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
7 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Wiedemann-Rautenstrauch
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Novel POLR3A Gene Mutation Results in Wiedemann-Rautenstrauch Syndrome With Striking Cutis Laxa and Myelofibrosis.
Wiedemann-Rautenstrauch syndrome is an extremely rare autosomal recessive progeroid disorder closely linked to mutations in POLR3A. Here, we report a case of a 4-year-old female patient carrying a novel compound-heterozygous variant in POLR3A. In addition to the classic Wiedemann-Rautenstrauch syndrome features-progressive diffuse alopecia, growth retardation, and abnormal white matter development-the patient presented with severe anemia and skin laxity, phenotypes not previously described in Wiedemann-Rautenstrauch syndrome. RT-qPCR analysis of skin tissue demonstrated a significant downregulation of POLR3A mRNA expression (p < 0.01). To our knowledge, this is the first report implicating an intronic POLR3A variant in Wiedemann-Rautenstrauch syndrome in the Chinese population, expanding both the mutational and phenotypic spectra of the disorder and underscoring its clinical heterogeneity.
POLR3A mutations cause nucleolus abnormalities and aberrant telomerase RNA metabolism in induced pluripotent stem cells from Wiedemann-Rautenstrauch premature aging syndrome patient.
Induced pluripotent stem cells (iPSCs) derived from patients with premature aging disorders are widely regarded as a foundation for both the study of fundamental aging mechanisms and preclinical testing of anti-aging therapies. The most well-studied is Hutchinson-Gilford progeria syndrome (HGPS), which is caused by a lamin A gene mutation. Comparing the progeroid phenotype in cell models of distinct premature aging syndromes is critical for identifying early and common aging hallmarks. In this study, using a non-integrative episomal approach we reprogrammed iPSCs from cells of a patient suffering from Wiedemann-Rautenstrauch Syndrome (WRS), which is caused by bi-allelic pathogenic mutations of the RNA polymerase III subunit A gene (POLR3A). In parallel, an iPSC line with the classic HGPS caused by a lamin A mutation was obtained. HGPS and WRS patient fibroblasts showed similar signs of cellular aging; however, unlike HGPS, the causal link between the premature aging phenotype and WRS driving mutations is unclear. RNA polymerase III is required for the transcription of small nuclear RNAs and being a target of TORC1 (Target of Rapamycin kinase Complex 1), it plays a role in longevity and aging in model organisms. Whereas lamin A is downregulated in iPSCs, allowing for regeneration of HGPS iPSCs, we found that POLR3A is upregulated during reprogramming. Enhanced expression of mutant POLR3A in WRS iPSCs led to nucleolus abnormalities and telomerase RNA component (TERC) sequestration in the nucleoli in WRS iPSCs. WRS iPSCs may be an important model for developing new therapeutic approaches affecting premature aging of stem cells.
Clinical and molecular insights into Wiedemann-Rautenstrauch syndrome: A case report and genetic analysis of the c.2707G>A variant in the POLR3A gene.
Wiedemann-Rautenstrauch syndrome (WRS) is a rare neonatal progeroid disorder primarily associated with pathogenic variants in POLR3A. However, the pathogenicity of certain variants remains unclear. Here, we report a WRS case carrying the POLR3A c.2707G > A (p.Gly903Arg) variant and explore its potential role in disease pathogenesis through in silico predictive and structural modeling analyses. Evolutionary conservation analysis, along with functional impact predictions from Provean, SIFT, PolyPhen-2, MutationTaster, MutPred2, Align GVGD, SNAP, and PhD-SNP, consistently classified the variant as deleterious. Splicing predictions using Human Splicing Finder (HSF) and SpliceAI suggested disruption of regulatory motifs and activation of a cryptic splice site. To find potential structural consequences, molecular modeling of the wild-type and mutant RNA polymerase III complex (PDB: 7DN3) was performed using PyMOL, while DynaMut analysis revealed destabilizing effects, decreased residue flexibility, and steric clashes that could impair complex function. By integrating genetic, computational, and structural approaches, this study not only provides a comprehensive characterization of the POLR3A c.2707G > A (p.Gly903Arg) variant but also contributes to the expanding spectrum of genetic findings in WRS. Our findings highlight the importance of case reports in refining genotype-phenotype correlations and emphasize the need for further functional validation to elucidate the molecular mechanisms underlying WRS.
Investigating telomere length in progeroid syndromes: implications for aging disorders.
Progeroid syndromes are rare genetic disorders that impact patients' health and lifespans and are characterized by symptoms that mimic the normal aging process. Telomere length is one of the aging hallmarks, a phenomenon linked to cellular aging. Telomere attrition was observed in different progeroid syndromes, such as Nijmegen breakage syndrome patients and Werner syndrome, indicating its contribution to the progeroid phenotype. However, whether it is a common feature in all progeroid syndromes is still unclear. Therefore, in this study, we aimed to estimate telomere length using the DNA methylation-based estimator of human telomere length in publicly available DNA methylation data from patients with Werner Syndrome, Hutchinson-Gilford Progeria Syndrome, Berardinelli-Seip Congenital Lipodystrophy type 2, and Dyskeratosis congenita, along with additional data provided by our laboratory from patients with Cerebroretinal Microangiopathy with Calcifications and Cysts and Wiedemann-Rautenstrauch Syndrome. Our findings revealed that certain progeroid syndromes, including classical Werner Syndrome, Berardinelli-Seip Congenital Lipodystrophy type 2, and Dyskeratosis congenita, have significant telomere attrition conversely to Hutchinson-Gilford Progeria Syndrome, Cerebroretinal Microangiopathy with Calcifications and Cysts, Wiedemann-Rautenstrauch Syndrome, and atypical Werner Syndrome. In conclusion, this study addresses a critical gap by providing new insights into the role of telomere attrition across different progeroid conditions. Further research is needed to elucidate the effect of telomere attrition on progeroid syndromes and its implications.
Prenatal and Postnatal Diagnosis and Genetic Background of Corpus Callosum Malformations and Neonatal Follow-Up.
The corpus callosum is one of the five main cerebral commissures. It is key to combining sensory and motor functions. Its structure can be pathological (dysgenesis) or completely absent (agenesis). The corpus callosum dys- or agenesis is a rare disease (1:4000 live births), but it can have serious mental effects. In our study, we processed the data of 64 pregnant women. They attended a prenatal diagnostic center and genetic counseling from 2005 to 2019 at the Department of Obstetrics and Gynecology at Semmelweis University. The pregnancies had the following outcomes: 52 ended in delivery, 1 in spontaneous abortion, and 11 in termination of pregnancy (TOP) cases (n = 64). The average time of detection with imaging tests was 25.24 gestational weeks. In 16 cases, prenatal magnetic resonance imaging (MRI) was performed. If the abnormality was detected before the 20th week, a genetic test was performed on an amniotic fluid sample obtained from a genetic amniocentesis. Karyotyping and cytogenetic tests were performed in 15 of the investigated cases. Karyotyping gave normal results in three cases (46,XX or XY). In one of these cases, postnatally chromosomal microarray (CMA) was later performed, which confirmed Aicardi syndrome (3q21.3-21.1 microdeletion). In one case, postnatally, the test found Wiedemann-Rautenstrauch syndrome. In other cases, it found X ring, Di George syndrome, 46,XY,del(13q)(q13q22) and 46,XX,del(5p)(p13) (Cri-du-chat syndrome). Edwards syndrome was diagnosed in six cases, and Patau syndrome in one case. We found that corpus callosum abnormalities are often linked to chromosomal problems. We recommend that a cytogenetic test be performed in all cases to rule out inherited diseases. Also, the long-term outcome does not just depend on the disease's severity and the associated other conditions, and hence proper follow-up and early development are also key. For this reason, close teamwork between neonatology, developmental neurology, and pediatric surgery is vital.
Publicações recentes
Novel POLR3A Gene Mutation Results in Wiedemann-Rautenstrauch Syndrome With Striking Cutis Laxa and Myelofibrosis.
POLR3A mutations cause nucleolus abnormalities and aberrant telomerase RNA metabolism in induced pluripotent stem cells from Wiedemann-Rautenstrauch premature aging syndrome patient.
Clinical and molecular insights into Wiedemann-Rautenstrauch syndrome: A case report and genetic analysis of the c.2707G>A variant in the POLR3A gene.
Investigating telomere length in progeroid syndromes: implications for aging disorders.
Prenatal and Postnatal Diagnosis and Genetic Background of Corpus Callosum Malformations and Neonatal Follow-Up.
📚 EuropePMC37 artigos no totalmostrando 30
Novel POLR3A Gene Mutation Results in Wiedemann-Rautenstrauch Syndrome With Striking Cutis Laxa and Myelofibrosis.
The Journal of dermatologyPOLR3A mutations cause nucleolus abnormalities and aberrant telomerase RNA metabolism in induced pluripotent stem cells from Wiedemann-Rautenstrauch premature aging syndrome patient.
BiogerontologyClinical and molecular insights into Wiedemann-Rautenstrauch syndrome: A case report and genetic analysis of the c.2707G>A variant in the POLR3A gene.
Experimental gerontologyInvestigating telomere length in progeroid syndromes: implications for aging disorders.
AgingPrenatal and Postnatal Diagnosis and Genetic Background of Corpus Callosum Malformations and Neonatal Follow-Up.
Children (Basel, Switzerland)[Wiedemann-Rautenstrauch syndrome. The first description of a clinical case in the Russian Federation].
Problemy endokrinologiiLoss-of-function variants affecting the STAGA complex component SUPT7L cause a developmental disorder with generalized lipodystrophy.
Human geneticsThe Genetic Basis of the First Patient with Wiedemann-Rautenstrauch Syndrome in the Russian Federation.
GenesFurther delineation of Wiedemann-Rautenstrauch syndrome linked with POLR3A.
Molecular genetics & genomic medicineBiallelic POLR3A variants cause Wiedemann-Rautenstrauch syndrome with atypical brain involvement.
Clinical and experimental pediatricsA synonymous variant contributes to a rare Wiedemann-Rautenstrauch syndrome complicated with mild anemia via affecting pre-mRNA splicing.
Frontiers in molecular neuroscienceA Case of Wiedemann-Rautenstrauch Syndrome With Fatal Hyperkalemic Renal Faliure.
CureusDistinguishing severe phenotypes associated with pathogenic variants in POLR3A.
American journal of medical genetics. Part AA novel homozygous synonymous variant further expands the phenotypic spectrum of POLR3A-related pathologies.
American journal of medical genetics. Part AWhole-exome sequencing reveals POLR3B variants associated with progeria-related Wiedemann-Rautenstrauch syndrome.
Italian journal of pediatricsWiedemann-Rautenstrauch syndrome in an Indian patient with biallelic pathogenic variants in POLR3A.
American journal of medical genetics. Part ATwo intronic cis-acting variants in both alleles of the POLR3A gene cause progressive spastic ataxia with hypodontia.
Clinical geneticsNucleolar disruption, activation of P53 and premature senescence in POLR3A-mutated Wiedemann-Rautenstrauch syndrome fibroblasts.
Mechanisms of ageing and developmentPathophysiology of premature aging characteristics in Mendelian progeroid disorders.
European journal of medical geneticsUnique combination and in silico modeling of biallelic POLR3A variants as a cause of Wiedemann-Rautenstrauch syndrome.
European journal of human genetics : EJHGA variant of neonatal progeroid syndrome, or Wiedemann-Rautenstrauch syndrome, is associated with a nonsense variant in POLR3GL.
European journal of human genetics : EJHGPOLR3A Identified as Major Locus for Autosomal Recessive Wiedemann-Rautenstrauch Syndrome: New findings show "compelling evidence" that POLR3A mutations underlie the etiology of autosomal-recessive WRS.
American journal of medical genetics. Part ABi-allelic POLR3A Loss-of-Function Variants Cause Autosomal-Recessive Wiedemann-Rautenstrauch Syndrome.
American journal of human geneticsSpecific combinations of biallelic POLR3A variants cause Wiedemann-Rautenstrauch syndrome.
Journal of medical geneticsRandom walk with restart on multiplex and heterogeneous biological networks.
Bioinformatics (Oxford, England)De Novo Mutations in SLC25A24 Cause a Craniosynostosis Syndrome with Hypertrichosis, Progeroid Appearance, and Mitochondrial Dysfunction.
American journal of human geneticsWiedemann-Rautenstrauch Syndrome With Bilateral Tarsal Kink: Three Sutures for Correction.
The Journal of craniofacial surgeryWiedemann-Rautenstrauch syndrome: A phenotype analysis.
American journal of medical genetics. Part ANeonatal progeriod syndrome associated with biallelic truncating variants in POLR3A.
American journal of medical genetics. Part AOphthalmic manifestations in a case of Wiedemann-Rautenstrauch syndrome.
Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and StrabismusAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Wiedemann-Rautenstrauch.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Wiedemann-Rautenstrauch
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Novel POLR3A Gene Mutation Results in Wiedemann-Rautenstrauch Syndrome With Striking Cutis Laxa and Myelofibrosis.
- POLR3A mutations cause nucleolus abnormalities and aberrant telomerase RNA metabolism in induced pluripotent stem cells from Wiedemann-Rautenstrauch premature aging syndrome patient.
- Clinical and molecular insights into Wiedemann-Rautenstrauch syndrome: A case report and genetic analysis of the c.2707G>A variant in the POLR3A gene.
- Investigating telomere length in progeroid syndromes: implications for aging disorders.
- Prenatal and Postnatal Diagnosis and Genetic Background of Corpus Callosum Malformations and Neonatal Follow-Up.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:3455(Orphanet)
- OMIM OMIM:264090(OMIM)
- MONDO:0009910(MONDO)
- GARD:330(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q3508774(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome Wiedemann-Rautenstrauch
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Reposicionamento
- fonte: Drug Repurposing Hub